Chromatographie: théorie
Bibliographie:
"Chimie organique expérimentale"
Auteurs: Chavanne, Beaudoin, Jullien, Flamand . Editeur: Belin
"Analyse chimique (méthodes et techniques instrumentales modernes)"
Auteur: Rouessac . Editeur: Masson
"144 manipulations de chimie générale et minérale"
Auteur: Defranceschi. Editeur : Ellipses
cours de chromatographie en phase gazeuse de Jean UMBER, professeur au lycee Louis Vincent.
1. Chromatographie: aspects généraux:
1.1. Définitions:
La chromatographie est une méthode physique de séparation basée sur les différences d'affinités des substances à analyser à l'égard de deux phases, l'une stationnaire ou fixe, l'autre mobile. Selon la technique chromatographique mise en jeu, la séparation des composants entraînés par la phase mobile, résulte soit de leur adsorption et de leur désorption successives sur la phase stationnaire, soit de leur solubilité différente dans chaque phase.
On définit un coefficient de partition K:
K =
On peut classer les méthodes chromatographiques d'après la nature des phases utilisées ou celle des phénomènes mis en oeuvre dans la séparation.
Nature des phases:
Phase fixe:
La phase fixe peut être solide ou liquide. Les solides, silice ou alumine traitées, permettent la séparation des composants des mélanges grâce à leurs propriétés adsorbantes. Ils peuvent être employés comme remplissage d'une colonne (chromatographie par gravité et chromatographie à haute performance ou HPLC) ou étalés en couche mince sur une plaque de verre, d'aluminium ou sur une feuille de matière plastique (chromatographie sur couche mince ou CCM)
La phase fixe peut aussi être constituée par un liquide imprégnant un support solide ou encore par une chaîne carbonée fixée sur un support (phase greffée). ainsi en chromatographie sur papier, la phase fixe est formée par l'eau que les molécules de cellulose du papier adsorbent, alors qu'en chromatographie en phase gazeuse, elle est constituée d'un liquide peu volatil et thermiquement stable imprégnant un granulé poreux.
Phase mobile:
La phase mobile est:
soit un gaz (ex: chromatographie en phase gazeuse): la phase mobile est appelée gaz vecteur ou gaz porteur.
soit un liquide (ex: chromatographie sur papier, couche mince ou colonne): la phase mobile est appelée éluant.
Nature des phénomènes:
On distingue quatre types de phénomènes que nous allons étudier successivement:
chromatographie d'adsorption
chromatographie de partage
chromatographie ionique
chromatographie d'exclusion
1.2. Chromatographie d'adsorption:
Elle est illustrée par la séparation chromatographique classique, sur colonne remplie ou sur couche mince, des composés moléculaires.
a

b
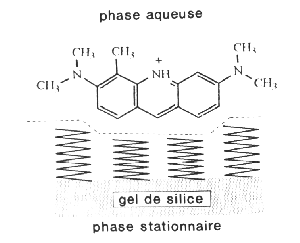
Phénomènes d'adsorption et de partage.
a) l'adsorption est un phénomène d'interface à la différence de l'absorption (reproduit avec l'autorisation de M. Laguës, L'Actualité Chimique 1990, (1) p.l7)
b) lorsqu'à la surface du gel de silice on greffe une monocouche d'hydrocarbure, les molécules, qui présentent une partie lipophile et l'autre hydrophile, s'orientent à l'interface comme en témoigne l'exemple de l'orangé d'acridine en phase aqueuse. (Chem. & Eng. News 1991, 69(37) p.25).
Polarité des phases:
Les séparations sont basées sur le principe de polarité c'est à dire l'existence de dipôles.
Phase mobile:
Le pouvoir éluant d'un liquide dépend de sa propre polarité. Les liquides classés ci-dessous le sont par polarité croissante. On obtient ainsi une série éluotropique.
éther de pétrole
cyclohexane
tétrachlorométhane
trichloréthène
toluène
benzène
dichlorométhane
éther diéthylique
trichlorométhane
éthanoate d'éthyle
pyridine
propanone
propan-1-ol
éthanol
méthanol
eau
acide éthanoïque
Adsorbants:
Les adsorbants figurant dans la liste ci-dessous sont classés selon l'ordre croissant de leurs forces d'interactions avec des composés polaires.
Papier, cellulose
Kieselguhr, terre de diatomées
Amidon
Sucres
Talc
Carbonate de sodium
Oxyde de magnésium
Gel de silice
Alumine
Charbon activé
Le gel de silice et l'alumine sont les adsorbants les plus utilisés. En général, plus un adsorbant est actif, plus il retient fortement les composés polaires. Il est cependant possible de traiter un adsorbant pour modifier ses capacités d'adsorption et ses propriétés: Plus la teneur en eau d'un adsorbant est faible (ce qui a pour conséquence la présence d'un plus grand nombre de sites d'adsorption pour le soluté), plus il est polaire ou actif.
Interactions entre le composé à analyser et les deux phases:
Lorsque les solutés sont neutres, l'ordre d'adsorption sur un adsorbant polaire comme le gel de silice ou l'alumine est le même que celui présenté pour les solvants. Les solutés apolaires (ex: alcanes) sont peu adsorbés alors que les solutés polaires (ex: méthanol) le sont fortement.
Par contre, on utilisera de préférence l'alumine pour séparer des solutés basiques comme les amines et le gel de silice pour les phénols et les acides car les solutés acides sont fortement adsorbés par l'alumine alors que les solutés basiques le sont par la silice.
1.3. Chromatographie de partage:
Elle est fondée sur la différence de solubilté des substances à séparer dans deux fluides parfaitement miscibles. Elle est mise en pratique en chromatographie sur papier. Un des fluides est un liquide retenu sur un support inerte et constitue la phase stationnaire. L'autre, liquide ou gaz en déplacement, constitue la phase mobile.Le facteur principal qui intervient est le coefficient de partage entre chaque phase.On peut séparer des solutés dont les coefficients de partage entre les deux phases sont différents. Les plus solubles dans la phase mobile se déplacent plus facilement que ceux qui le sont moins.
Un autre facteur qui intervient est la polarité de la phase: ainsi en HPLC on peut utiliser des phases stationnaires peu ou non polaires, la phase mobile étant polaire (eau ou mélange eau - méthanol): on parlera alors de chromatographie de partage à polarité de phase inversée.
1.4. Chromatographie ionique:
La phase mobile est une solution tampon aqueuse et la phase stationnaire la plus courante est constituée de polystyrène sous forme de sphères de quelques micromètres de diamètre, lesquelles ont été chimiquement transformées en surface pour faire apparaître des sites ioniques. Ces phases permettent l'échange de leurs contre-ions mobiles avec des ions, de même signe, présents dans la phase mobile. La séparation repose sur les coefficients de distribution ionique entre les deux phases.
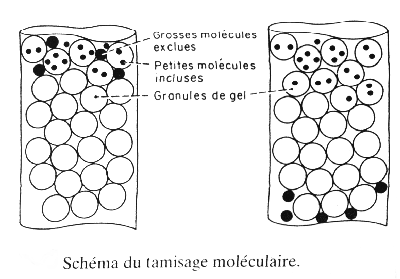
1.5. Chromatographie d'exclusion:
La phase stationnaire est généralement un polymère poreux dont les pores ont des dimensions choisies en rapport avec la taille des espèces à séparer. On réalise une sorte de tamis à l'échelle moléculaire, dit à perméation sélective. Le coefficient de partition s'appelle dans ce cas le coefficient de diffusion. Cette technique est encore appelée filtration sur gel ou perméation de gel selon la nature de la phase mobile (aqueuse ou organique).
Le diamètre des pores est une caractéristique de chaque type de gel.
Un mélange de solutés de masses molaires variables traverse une épaisseur donnée de gel: les grosses molécules, celles dont le diamètre est supérieur à celui des pores, sont exclues et éluées les premières; les petites et moyennes molécules sont éluées plus tardivement, car incluses, leur migration est freinée en diffusant dans le gel.
La séparation est donc réalisée par le fait que les solutés sont élués dans l'ordre inverse des masses molaires.
2 Chromatographie sur couche mince (CCM)
2.1.Définition et appareillage:
La chromatographie sur couche mince (CCM) repose principalement sur des phénomènes d'adsorption : la phase mobile est un solvant ou un mélange de solvants, qui progresse le long d'une phase stationnaire fixée sur une plaque de verre ou sur une feuille semi-rigide de matière plastique ou d'aluminium. Après que l'échantillon ait été déposé sur la phase stationnaire, les substances migrent à une vitesse qui dépend de leur nature et de celle du solvant.
Les principaux éléments d'une séparation chromatographique sur couche
mince sont:
la cuve chromatographique : un récipient habituellement en verre, de forme variable, fermé par un couvercle étanche.
la phase stationnaire : une couche d'environ 0,25 mm de gel de silice ou d'un autre adsobant est fixée sur une plaque de verre à l'aide d'un liant comme le sulfate de calcium hydraté (plâtre de Paris) l'amidon ou un polymère organique.
l'échantillon : environ un microlitre (µl) de solution diluée ( 2 à 5 %) du mélange à analyser, déposé en un point repère situé au-dessus de la surface de l'éluant.
l'éluant : un solvant pur ou un mélange : il migre lentement le long de la plaque en entraînant les composants de l'échantillon.
2.2.Principe de la technique.
Lorsque la plaque sur laquelle on a déposé l'échantillon est placée dans la cuve, l'éluant monte à travers la phase stationnaire, essentiellement par capillarité. En outre, chaque composant de l'échantillon se déplace à sa propre vitesse derrière le front du solvant. Cette vitesse dépend d'une part, des forces électrostatiques retenant le composant sur la plaque stationnaire et, d'autre part, de sa solubilité dans la phase mobile. Les composés se déplacent donc alternativement de la phase stationnaire à la phase mobile, l'action de rétention de la phase stationnaire étant principalement contrôlée par des phénomènes d'adsorption. Généralement, en chromatographie sur couche mince, les substances de faible polarité migrent plus rapidement que les composants polaires.
2.3.Applications de la CCM.
Lorque les conditions opératoires sont connues, elle permet un contrôle aisé et rapide de la pureté d'un composé organique. Si l'analyse, réalisée avec divers solvants et différents adsorbants, révèle la présence d'une seule substance, on peut alors considérer que cet échantillon est probablement pur.
De plus, étant donné que la chromatographie sur couche mince indique le nombre de composants d'un mélange, on peut l'employer pour suivre la progression d'une réaction.
La chromatographie sur couche mince est également la technique habituellement employée pour rechercher le meilleur solvant, avant d'entreprendre une séparation par chromatographie sur colonne.
2.4.Adsorbants et plaques chromatographiques.
Par ordre d'importance décroissante, les adsorbants employés en CCM sont : le gel de silice, l'alumine, le kieselguhr et la cellulose.
Les plaques vous seront fournies prêtes à l'emploi.
2.5.Choix de l'éluant.
L'éluant est formé d'un solvant unique ou d'un mélange de solvants.
Un éluant qui entraîne tous les composants de l'échantillon est trop polaire; celui qui empêche leur migration ne l'est pas suffisamment.
Une méthode simple pour trouver l'éluant approprié consiste à préparer des solutions de l'échantillon dans différents solvants, en concentration d'environ 2 à 5% en volume. A l'aide d'une micropipette, on dépose une goutte de chaque solution sur une plaque, chacune séparée d'environ 1 cm. Le meilleur éluant est celui qui, lorsqu'il a terminé sa migration, a entraîné le soluté à une distance d'environ la moitié de celle qu'il a parcourue.
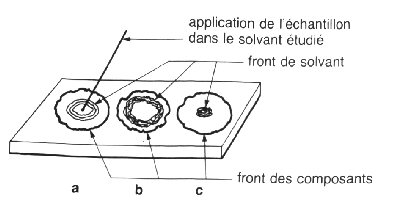
Une autre méthode consiste à déposer une solution des substances à analyser en plusieurs points, séparés d'environ 2 cm. Après séchage, on applique au centre de chaque point une micropipette remplie de solvant; Après diffusion, l'éluant qui convient sépare les solutés.
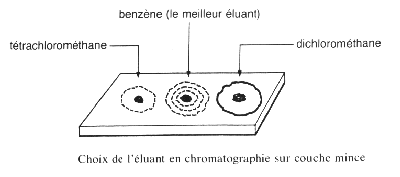
Choix de l'éluant dans le cas d'analyses :
d'hydrocarbures : hexane, éther de pétrole ou benzène.
de groupements fonctionnels courants : hexane ou éther de pétrole mélangés en proportions variables avec du benzène ou de l'éther diéthylique forment un éluant de polarité moyenne.
de composés polaires : éthanoate d'éthyle, propanone ou méthanol.
2.6.Dépôt de l'échantillon.
L'échantillon est mis en solution (2 à 5 %) dans un solvant volatil, qui n'est pas forcément le même que l'éluant : on emploie fréquemment le trichlorométhane (chloroforme),la propanone ou le dichlorométhane.La solution est déposée en un point de la plaque situé à environ 1 cm de la partie inférieure.
Il est important que le diamètre de la tache produite au moment du dépôt soit faible; idéalement, il ne devrait pas dépasser 3 mm. Ce sont généralement les dépôts les moins étalés qui permettent les meilleures séparations. Pour augmenter la quantité déposée, il est toujours préférable d'effectuer plusieurs dépôts au même point, en séchant rapidement entre chaque application plutôt que de déposer en une seule fois un grand volume d'échantillon qui produirait une tache plus large.
L'échantillon est déposé à l'aide d'une micropipette ou d'un tube capillaire en appuyant légèrement et brièvement l'extrémité de la pipette sur la couche d'adsorbant en prenant soin de ne pas le détériorer. On peut aussi utiliser l'extrêmité, un peu émoussée, d'un cure-dent.
On vérifie l'identité des composants présumés d'un échantillon, en procédant à un dépôt séparé d'une solution de chacun d'eux puis à celui de leur mélange. Ces solutions témoins permettent de comparer la migration de chaque composé avec celle de l'échantillon à analyser.
2.7.Développement de la plaque.
Le développement consiste à faire migrer le solvant sur la plaque. Dans les analyses usuelles de laboratoire, le principal type de développement est la chromatographie ascendante : la plaque est placée en position verticale dans une cuve et le solvant qui en recouvre le fond monte par capillarité.
Le niveau de liquide est ajusté à environ 0,5 cm du fond de la cuve; on place souvent du papier filtre contre les parois de la cuve pour saturer plus rapidement la cuve en vapeurs d'éluant et éviter les effets de bords.Pendant le développement du chromatogramme, la cuve doit demeurer fermée et ne pas être déplacée.
Lorsque la position du front du solvant arrive à environ 1 cm de l'extrémité supérieure, la plaque est retirée de la cuve, le niveau atteint par le solvant est marqué par un trait fin, puis la plaque est séchée à l'air libre ou à l'aide d'un séchoir.
2.8.Révélation.
L'identification des substances isolées se fait selon différentes méthodes (valable également pour la chromatographie sur papier):
directement si les substances sont colorées
à l'aide de révélateurs si elles sont incolores afin de les transformer en taches colorées; les produits sont souvent décelés par leurs réactions fonctionnelles classiques: les acides aminés par la ninhydrine qui donne avec la plupart une couleur bleu-violet, les acides organiques par des indicateurs colorés, les sucres par le réactifde Molisch qui utilise le pouvoir réducteur des sucres. Quelques réactifs comme l'iode ou le permanganate donnent des colorations non spécifiques avec la plupart des composés organiques.
toutes les ubstances ayant une absorption dans la région au-dessus de 230 nm sont étudiées sur des supports additionnnés de corps fluorescents par irradiation de lumière UV à ondes courtes (lmax< 254 nm). L'emploi de couches non additionnées de produits fluorescents permet aussi la mise en évidence de beaucoup de substances dans l'UV à ondes courtes (lmax <254 nm) ou à ondes lon,gues (lmax > 366 nm) par suite de la fluorescence propre des composés.
Dans tous les cas, il faut noter les positions des taches colorées juste à la fin de la chromatographie en les cerclant car certains produits disparaissent avec le temps.
2.9.Calcul de Rf (retarding factor ou rapport frontal)
di : distance parcourue par le composé (mesure au centre de la tache)
ds : distance parcourue par le front du solvant
Pour un couple éluant et support déterminé, Rf est une caractéristique de chaque soluté à la température de l'expérience. Rf est toujours indépendant dfe la longueur de bande utilisée.
2.10.Description d'une analyse par CCM selon l'ordre chronologique.
Préparation de la cuve chromatographique.
Introduire l'éluant ou le mélange de solvants.
Ajuster le niveau à environ 0,5 cm du fond de la cuve.
Fermer le récipient (la cuve doit être saturée de vapeur de solvant). Pour que la saturation et l'élution soient plus rapides, on peut placer une bande de papier filtre contre les parois de la cuve chromatographique.
Dépôt de l'échantillon sur la plaque.
Procéder au nettoyage de la plaque si nécessaire.
Dissoudre l'échantillon dans un solvant approprié en solution de 2 à 5 % .
Déposer environ 0,5 µl de la solution en un point situé à 1 cm de l'extrémité inférieure de la plaque; le diamètre de la tache doit être d'environ 2 mm pour la disposition de plusieurs produits.
Sécher à l'aide d'un séchoir;éventuellement faire de nouvelles applications
Développement du chromatogramme.
Placer la plaque dans la cuve en position verticale.
Refermer le récipient.
Lorsque le front du solvant se trouve à environ 1 cm de l'extrémité supérieure de la plaque, la retirer et marquer cette position.(le trait peut être tracé à l'avance et servir de repère pour arrêter l'élution).
Révélation et calcul de Rf
Sécher la plaque à l'aide d'un séchoir
Révéler les taches sous une lampe U V ou à l'aide d'un révélateur
Cercler les taches et pointer leur centre.
Calculer les Rf
3. Chromatographie sur papier:
3.1. Principe de la technique et applications:
La technique ressemble à celle de la CCM mais le principe repose sur des phénomènes de partage. La phase mobile est le plus souvent un solvant organique et l'eau; la phase stastionnaire est constituée par l'eau elle-même adsorbée sur la cellulose du papier ou liée chimiquement à elle. Comme en chromatographie sur couche mince, l'échantillon, mis en solution, est déposé en un point repère du papier et le solvant, qui se déplace par capillarité, fait migrer les composants de l'échantillon à des vitesses variables selon leur solubilité. Généralement, les composés les plus solubles dans l'eau ou ceux qui forment facilement des associations par liaisons hydrogène sont fortement retenus par la phase stationnaire et migrent donc lentement.
Lorsque l'eau est un des solvants de la phase mobile, le ou les solvants organiques doivent y être assez solubles. Des produits comme l'acide éthanoïque, le propanol, le phénol ou la pyridine sont les solvants les plus fréquemment utilisés en mélange avec de l'eau pour développer un chromatogramme.
La chromatographie sur papier est employée principalement pour l'analyse de composés très polaires, tels les acides aminés, les sucres et les composés polyfonctionnels.
Ses plus grands inconvénients par rapport à la CCM sont:
une durée de développement beaucoup plus longue
une séparation généralement moins bonne.
3.2. Papier:
On peut utiliser du papier filtre ordinaire, mais il est préférable de se procurer du papier conçu pour cet usage, ayant un faible taux d'impuretés et dont les caractéristiques sont uniformes. Les marques principales sont Whatman, Schleicher et Schüll, Durieux, Arches. Il existe huit catégories de papier Whatman, classés selon leur épaisseur, la texture de leur surface et la vitesse avec laquelle l'eau y diffuse. Par exemple le papier Whatman n° 1 est le plus utilisé, mais si on désire une grande vitesse d'écoulement, on emploiera le n° 4; le papier n° 20 est très lent, mais il permet une meilleure séparation , donnant des taches très denses et uniformes.
La description de l'analyse par chromatographie sur papier est identique à celle sur couche mince.
4. Chromatographie sur colonne:
Alors que les autres méthodes chromatographiques sont habituellement employées pour l'analyse et la séparation de très faibles quantités de produits, la chromatographie sur colonne peut être une méthode préparative; elle permet en effet la séparation des constituants d'un mélange et leur isolement , à partir d'échantillons dont la masse peut atteindre plusieurs grammes.
Elle présente cependant plusieurs inconvénients:
de grandes quantités de solvant sont nécessaires à l'élution
la durée de l'élution est généralemnt très grande
la détection des composés exige une attention constante.
Elle est adaptée à la purification de faibles quantités de produit, lorsque les conditions opératoires sont au point. Cependant, la méthode étant très empirique, sa mise au point nécessite souvent de nombreux essais.
4.1. Description et principe:
C'est une technique basée sur des phénomènes d'adsorption. La phase solide, le plus souvent l'alumine ou la silice, remplit une colonne de longueur et de section variables; l'échantillon, en solution concentrée, est déposé en haut de la colonne et la séparation des composants résulte de l'écoulement continu d'un éluant, traversant la colonne par gravité ou sous l'effet d'une faible pression. On peut utiliser comme éluant un solvant unique ou bien accroître progressivement la polarité de l'éluant de façon à accélérer le déplacement des composés.
Les molécules sont entraînées vers le bas à des vitesses variables selon leur affinité pour l'adsorbant et leur solubilité dans l'éluant. Le chromatogramme se développe en formant une succession de zones cylindriques qui se séparent en migrant vers le bas. A mesure que chaque zone s'écoule de la colonne, on la recueille.
4.2. Facteurs dont dépend la séparation:
Quatre facteurs interviennent:
l'adsorbant
l'éluant
la dimension de la colonne
la vitesse d'élution
Adsorbant:
Le plus utilisé est l'alumine; cependant, on la limitera aux composés organiques stables car, sous sa forme basique, elle peut provoquer la déshydratation des esters par exemple.
Le gel de silice est également fréquemment utilisé pour la séparation de composés qui n'ont pas une stabilité suffisante pour être traités par l'alumine.
La granulométrie de l'adsorbant doit être supérieure à celle des adsorbants utilisés en CCM. Leur taille est habituellement comprise entre 50 et 200 µm.
La quantité d'adsorbant dépend de la difficulté de la séparation et de la masse d'échantillon.On peut considérer que pour chaque gramme d'échantillon, il faut 30 à 50 g d'adsorbant si la polarité des composants à séparer est très différente et jusqu'à 200 g si la séparation est difficile.
Eluant:
L'éluant est en général un mélange de deux solvants. Au début de l'élution, on commence par le solvant le moins polaire qui entraîne les substances les moins retenues par l'adsorbant (les moins polaires). Ensuite on fait varier la composition de l'éluant en additionnant graduellement le solvant le plus polaire. Ainsi les composés les plus polaires, retenus sur l'adsorbant, ne migreront que graduellement vers le bas de la colonne.
Dimension de la colonne:
Les colonnes spécialement conçues pour cet usage ont à leur base une plaque de verre fritté ou de porcelaine qui permet l'écoulement libre de l'éluant tout en empêchant le passage de l'adsorbant. On peut aussi utiliser une burette, au fond de laquelle on place un tampon de laine de verre et du sable.
La quantité d'adsorbant est telle qu'il occupe une hauteur égale à environ 10 fois le diamètre de la colonne. Il faut également prévoir un espace de 10 cm environ au-dessus de l'adsorbant pour placer le solvant.
Vitesse d'élution:
Elle doit être la plus constante possible. Il faut qu'elle soit suffisamment lente pour que le soluté soit au plus près de l'équilibre entre les phases liquide et adsorbée. Elle ne doit pas être trop lente car sinon les substances diffusent dans le solvant et on obtient des bandes de plus en plus larges et une séparation médiocre.
4.3. Remplissage de la colonne:
C'est l'opération le plus délicate car le remplissage doit être le plus homogène possible et exempt de bulle d'air. Les surfaces inférieure et supérieure de l'adsorbant doivent être parfaitement horizontales. La colonne étant verticale, le remplissage peut être réalisé selon deux méthodes au choix.
Remplissage par voie humide:
On prépare dans un bécher un mélange homogénéisé de l'adsorbant et du moins polaire des solvants utilisé pour le développement en ajoutant par petites quantités l'adsorbant dans le solvant pour obtenir une bouillie suffisamment fluide pour couler facilement.
A l'aide d'un entonnoir, on verse suffisamment de bouillie pour que l'adsorbant qui se dépose progressivement forme une couche d'environ 2 cm. On tapote les parois de la colonne pour favoriser le tassement de l'adsorbant. On ouvre alors le robinet pour que le solvant s'écoule lentement et on poursuit l'addition de la bouillie homogénéisée par portions successives. Quand tout l'adsorbant est introduit, on laisse décanter jusqu'à ce que le liquide qui surnage soit limpide.
Pendant l'opération,on doit veiller à ce que le niveau de solvant soit toujours supérieur à celui de l'adsorbant.
Remplissage par voie sèche:
La colonne est remplie au deux tiers par le moins polaire des deux solvants et l'adsorbant en poudre est ajouté en portions successives dans la colonne à l'aide d'un entonnoir; pendant l'addition, on frappe continuellement sur les parois pour obtenir un tassement maximal. Quand la première portion forme une couche d'environ 2 cm, on ouvre le robinet pour faire couler lentement le solvant. On termine comme précédemment.
4.4. Dépôt des produits à analyser:
Ils doivent former une zone cylindrique étroite dans le haut de la colonne.
Un liquide est déposé tel quel. Un solide sera dissous dans le minimum du moins polaire des deux solvants .
On ajuste d'abord le niveau de solvant pour qu'il soit juste au-dessus de celui de l'adsorbant. A l'aide d'une pipette, on coule l'échantillon au sommet de la colonne de façon uniforme sur toute la surface de la colonne sans la déformer. Si nécessaire, on ajuste à nouveau, comme précédemment, le niveau de liquide de la colonne : l'échantillon est ainsi adsorbé uniformément au sommet de la colonne. On peut placer un verre fritté ou une rondelle de papier filtre au-dessus de l'adsorbant pour prévenir une remise en suspension de l'adsorbant.
4.5. Elution:
On peut alimenter la colonne en continu à l'aide d'une ampoule de coulée ou bien ajouter manuellement l'éluant. Dans tous les cas, la surface de l'adsorbant ne doit jamais être au contact de l'air. En quelques minutes, une colonne laissée à sec se détériore: des fissures apparaissent dans la phase fixe et toute élution ultérieure se transforme en ruissellement.
Pour la plupart des opérations, une vitesse de 5 à 50 gouttes à la minute convient (la limite inférieure correspond aux séparations difficiles)
Lorsque l'analyse des fractions est terminée, on réunit celles qui correspondent à des produits identiques, en prenant soin d'éliminer celles qui correspondent à des recouvrements de zones. Les substances obtenues de cette façon sont généralement d'une très grande pureté.
5. Chromatographie en phase gazeuse (CPG):
Le principe de la séparation par C.P.G. consiste à partager l'échantillon à analyser entre deux phases. L'une de ces phases est un liquide stationnaire uniformément réparti sous forme d'une pellicule mince sur un solide inerte de grande surface spécifique, tandis que l'autre phase est un gaz mobile qui s'écoule à travers l'ensemble stationnaire.
5.1.Description d'un chromatographe.

Un chromatographe est constitué en première approximation de trois organes essentiels :
l'injecteur, le détecteur, la colonne
Injecteur:
Il permet d'introduire un liquide qui doit être vaporisé instantanément avant d'être transféré dans la colonne. Sa température doit être supérieure d'environ 20°C à la température du produit le moins volatil.
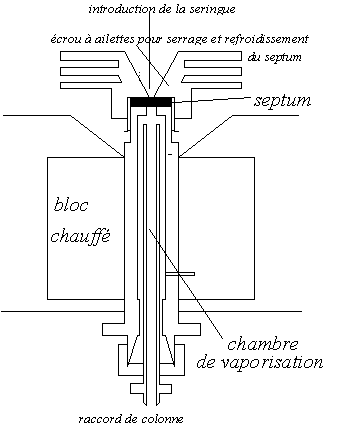
La figure suivante le représente: le gaz porteur, de préférence préchauffé, entre dans une chambre chauffée, obturée par une pastille d’élastomère, le septum, qui assure l’étanchéité. A l’aide d’une seringue hypodermique de petite capacité, on pique au travers du septum, afin que l’extrémité de l’aiguille arrive au-dessous du niveau de l’arrivée du gaz porteur, puis on pousse le piston pour réaliser l’injection.
|
|
Il faut que la chambre d’injection ait un volume aussi petit que possible, pour limiter les volumes morts du chromatographe.
D'autre part,si on observe des baisses de pression dans le circuit gazeux, cela indique souvent qu’il faut changer le septum, usé par les multiples injections. Il ne faut pas oublier de nettoyer la seringue d’injection avec un solvant volatil après chaque injection, puis de la sécher convenablement. |
Détecteur:
Il permet de mettre en évidence le passage des différents
gaz séparés par la colonne. La détection peut être basée sur des techniques de
mesures différentes. Le détecteur le plus utilisé en CPG est celui à
conductibilité thermique appelé catharomètre. (cas de notre chromatographe)
Sa température est généralement la même que celle de l'injecteur.
Catharomètre
Le catharomètre est un appareil simple et robuste, à réponse universelle, mais relativement peu sensible. Il est fondé sur une comparaison continuelle entre le flux de chaleur emporté par le gaz vecteur pur et le flux de chaleur emporté par le gaz vecteur chargé des molécules de soluté. Ces flux de chaleurs sont produits par des thermistances, parcourues par un courant continu de tension fixe, dans une enceinte thermostatée avec précision.
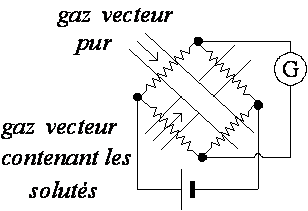
En faisant passer le gaz vecteur contenant les constituants séparés sur la cellule du détecteur, nous obtiendrons un signal chaque fois que l'un des constituants se présentera dans la cellule car la conductibilité thermique du gaz vecteur (qui traverse le détecteur) varie quand le constituant X traverse le détecteur.
L'élément sensible du capteur est un fil de platine parcouru par un courant constant
(I » 1200 mA). Quand l'équilibre entre l'apport d'énergie par effet Joule et la dissipation d'énergie par conduction et rayonnement est atteint, le fil prend une température d'équilibre qui est fonction de la conductibilité thermique du milieu gazeux où est placé le fil. Le passage d'un constituant différent dans le détecteur va modifier cette conductibilité, d'où une variation de la température du fil et par conséquent de sa résistance électrique.
L'élément sensible est incorporé dans l'une des branches d'un pont de Wheatstone. Dans l'autre branche est placé un fil absolument identique, mais qui est toujours placé dans le gaz porteur pur et dont la résistance sera donc constante; on constitue ainsi deux cellules, l'une de mesure et l'autre de référence.
Le pont est mis à l'équilibre en absence de soluté injecté.
Quand un constituant passe dans la cellule de mesure (porté par le gaz vecteur), un déséquilibre apparait. Il est amplifié et mesuré par l'enregistreur dont la déviation est proportionnelle à la différence de potentiel apparue.
Pour obtenir la plus grande réponse pour un soluté donné, il est donc nécessaire qu’il y ait la plus grande différence possible entre la conductivité de ce soluté et celle du gaz porteur. A cet effet, on utilise pratiquement toujours l’hydrogène (danger d’explosion) ou l’hélium comme gaz vecteur.
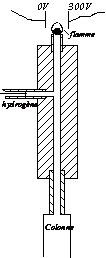
Détecteur à ionisation de flamme.
|
|
C’est un détecteur beaucoup plus sensible que le catharomètre, mais moins universel, car il ne donne de réponse qu’aux composés organiques. Il a aussi l’inconvénient, contrairement au catharomètre, de détruire le soluté qui le traverse, car son principe est de brûler, dans une flamme d’hydrogène, l’effluent apporté par de l’azote (gaz vecteur). Sous l’effet d’un champ électrostatique, il se forme des ions carbone de charge positive qui sont précipités sur une électrode où ils créent un courant d’ionisation que l’on amplifie grâce à un électromètre amplificateur. Sur un enregistreur, on obtient par conséquent un signal proportionnel au débit-masse du soluté dans le détecteur. En fait, il n’est pas exactement proportionnel au nombre d’atomes de carbone du composé concerné, car il y a une influence défavorable des autres atomes que C et H. Par contre, il est inutile de placer ce détecteur dans une enceinte thermostatée. |
Détecteur à capture d’électrons.
Une source telle que le tritium ![]() ou le
ou le ![]() envoie des
électrons libres dans le détecteur. Quand ce détecteur est traversé par des
substances ayant une affinité pour les électrons libres, il se produit des ions
qui, comme pour le détecteur à ionisation de flamme, dans le champ
électrostatique existant, sont recueillis par une électrode et forment un
courant d’ionisation à amplifier convenablement.
envoie des
électrons libres dans le détecteur. Quand ce détecteur est traversé par des
substances ayant une affinité pour les électrons libres, il se produit des ions
qui, comme pour le détecteur à ionisation de flamme, dans le champ
électrostatique existant, sont recueillis par une électrode et forment un
courant d’ionisation à amplifier convenablement.
Performances des détecteurs
|
Catharomètre |
Détecteur à ionisation de flamme |
Détecteur à capture d’électrons |
|
1 à 10 ng (tous composés) |
20 à 100 pg (composés organiques) |
0,1 pg (composés halogénés) |
Colonne:
C'est l'organe principal. Elle est constituée d'un tube généralement métallique de diamètre intérieur de l'ordre du millimètre. Ce tube contient la phase stationnaire constituée par un liquide adsorbant fixé sur un solide inerte ( ex : brique pilée, alumine etc... soigneusement calibrée)
On distingue les colonnes à remplissage proprement dit, constituées d’une tubulure en verre, acier ou autre métal (les plus fréquentes sont en acier inoxydables), dont les dimensions varient de 2 à 6 mm pour le diamètre intérieur et de 1 à 10 m pour la longueur. Le support remplissant la colonne est constitué de grains dont les dimensions varient de 60 à 70 µm; ils sont à base soit de matériau réfractaire soit de silice. La phase stationnaire est un liquide peu volatil, formant environ 10% de la masse du support non imprégné.
Par ailleurs, on utilise des colonnes capillaires, formées d’un tube de métal, de verre, de silice fondue ou de quartz, dont le diamètre intérieur est de l’ordre de 0,2 à 0,5 mm et la longueur de 50 à 100 m, ou davantage. L’adsorbant y est fixé sous forme d’une fine couche collée à la paroi du tube, ou bien la phase stationnaire est fixée en film mince, sans support, sur cette même paroi. Dans tous les cas, ces colonnes comportent un canal central largement ouvert, offrant peu de pertes de charge à la progression du gaz porteur.
La réussite d'une bonne séparation chromatographique dépend dans une large mesure du choix de la phase stationnaire.
On distingue les phases apolaires et les phases polaires. Les premières sont à base d’hydrocarbures aliphatiques saturés ou de silicones (squalane, apiezon,...). Les secondes sont des polymères possédant des fonctions polaires: polyols, polyesters, polyamides.
En général, les phases polaires retiennent plus les composés polaires, alors que ceux-ci sortent plus rapidement des colonnes apolaires que les composés du même nom.
Les adsorbants les plus classiques sont les adsorbants minéraux, tels le charbon actif, l’alumine, les tamis moléculaires. Ils sont pratiquement indispensables pour l’analyse des gaz, car ceux-ci sont peu solubles dans les phases stationnaires, et donc mal séparés par elles. Cependant, la désorption de ces gaz sur les adsorbants est lente, ce qui provoque généralement des traînées des pics.
On utilise aussi des adsorbants organiques à haut poids moléculaire. Ce sont des copolymères (du type styrène + divinylbenzène). Ils ont l’avantage de permettre toutes sortes d’analyses
![]()
Performances des colonnes.
Une colonne à remplissage bien préparée peut atteindre une efficacité de l’ordre de 1 500 plateaux théoriques par mètre, soit HEPT = 0,66 mm (Voir cette notion dans l'annexe I). Les colonnes capillaires atteignent facilement 2 000 à 10 000 plateaux théoriques par mètre, soit HEPT compris entre 0,5 et 0,1 mm.
On ne doit jamais effectuer d’analyse CPG sans avoir stabilisé l’ensemble de l’appareil en débit de gaz vecteur et en température pendant au moins deux heures.
En dehors des périodes d’utilisation, les colonnes doivent être bouchées pour éviter l’humidité pouvant se solubiliser dans la phase stationnaire, ainsi que l’oxydation de celle-ci.
La température de la colonne est en général inférieure de 20°C à celle du point d'ébullition du soluté le plus volatil. Plus la température de colonne est basse, meilleure est la séparation, mais cela risque d'allonger le temps d'analyse.
Le choix de la phase stationnaire conditionne la bonne séparation des constituants.Il faudra la choisir polaire ou apolaire en fonction de la nature des substances à séparer.
Le gaz employé (phase mobile) est un gaz inerte (hélium ou azote). Le gaz utilisé par notre installation est l'hélium. Ce gaz vecteur ou gaz porteur pousse les constituants à travers la colonne. En chaque point de cette dernière, il se produit un équilibre entre la fraction du constituant en phase stationnaire et en phase mobile. Il s'agit de chromatographie de partage.
L'ensemble des organes décrits ci-dessus est placé dans des enceintes thermostatées à des températures programmées selon la disposition des organes et la nature de l'échantillon à analyser.
5.2. Analyse qualitative en CPG:
Généralités:
Si on injecte un mélange de plusieurs liquides dans la colonne par l'intermédiaire de l'injecteur, ces liquides sont transformés en gaz, lesquels sont entraînés dans la colonne par le gaz vecteur. La phase stationnaire selon sa constitution, va plus ou moins retenir sélectivement chacun des produits. Les vitesses de progression seront différentes pour chaque constituant. Nous arriverons ainsi à éluer le mélange, c'est à dire à séparer dans l'espace et dans le temps les différents composants du mélange.
On appelle coefficient de partage K pour un constituant X :
K =
Ce coefficient de partage est un des paramètres qui conditionne la durée de parcours de la colonne par le constituant.
Si les autres paramètres (température, débit gazeux) sont constants, alors pour un mélange à analyser, la durée de parcours sera différente pour chaque constituant si leurs coefficients de partage sont différents.
Cette durée est appelée temps de rétention.
Moyennant un étalonnage préalable avec des produits purs, la CPG permet donc l'analyse qualitative des constituants d'un mélange.
Allure du chromatogramme
Un chromatogramme correct est composé de pics de forme symétrique, pas trop larges et bien séparés.
C'est en jouant sur les conditions opératoires que l'on arrive à un tel chromatogramme.
Les facteurs favorables à une bonne séparation sont :
des temps de rétention suffisamment différents (choix de la colonne)
des pics peu élargis.
Différence entre temps de rétention.
Le principal paramètre est la différence entre les coefficients de partage de chaque constituant. Ce dernier dépend beaucoup de la température et de la longueur de la colonne.
Une diminution de la température du four entraîne une augmentation du temps de rétention.
Une augmentation de la longueur de la colonne augmente les temps de rétention mais s'accompagne souvent d'un élargissement des pics.
Largeur des pics
L'obtention de pics larges est due au fait que les multiples équilibres de répartition des constituants entre les deux phases (liquide et gazeuse) durant leur séjour dans la colonne, au lieu de s'établir sur une longueur extrêment faible de la colonne s'établissent en fait sur une longueur non négligeable que l'on appelle plateau théorique.
5.3. Analyse quantitative en C.P.G:
Une fois identifié(s) le (ou les) soluté(s) intéressant(s), le chromatogramme permet aussi une analyse quantitative grâce à la relation :
|
mi= Ki Ai
|
mi : masse du soluté i injecté Ai : aire du pic représentant ce soluté Ki : coefficient de proportionnalité |
Il est donc nécessaire de déterminer pour chaque soluté la valeur de Ki
Ki dépend en outre du débit gazeux, de la température du détecteur ainsi que de l'intensité du courant qui le traverse.
Mesure de l’aire des pics.
On utilise essentiellement la triangulation manuelle et l’intégration automatique. (c'est cette dernière méthode qui sera utilisée ici.)
Quand les pics sont très pointus et très étroits, on peut se contenter des mesures des hauteurs H , alors proportionnelles aux aires.
Détermination du coefficient de proportionnalité.
Il est impossible avec les chromatographes courants de calculer le coefficient de proportionnalité par mesure directe de l’aire du pic enregistré quand on introduit une masse exacte, connue, d’un soluté d’un injecteur. Les seringues d’injection ne permettent pas de repérer le volume d’échantillon avec une précision suffisante. On aura donc recours à des méthodes d’étalonnage, qui, comme en analyse qualitative, feront de la chromatographie quantitative un procédé relatif vis-à-vis de substances connues. Voici les principales méthodes utilisées.
Normalisation interne.
On considère ici, en première approximation, que tous les Ki sont égaux (principalement dans les séries homologues telles que alcanes, alcools, etc..). On obtient alors les pourcentages en masse de chaque soluté de la manière suivante:
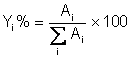
Méthode des ajouts dosés.
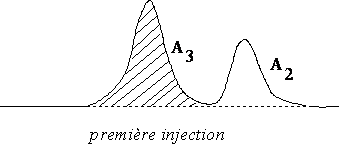
Le principe est le suivant: on prend d’abord le chromatogramme du mélange de solutés à étudier. Admettons que l’on cherche à déterminer le pourcentage en masse du composé n° 2. On va obtenir l’aire du pic correspondant A2 , et l’on détermine également celle d’un pic voisin, par exemple A3. On pèse ensuite exactement une masse M de ce mélange voisine de 1g par exemple. Puis un y rajoute une masse m0 du composé n°2, connue exactement, voisine de 300 mg environ. Le chromatogramme du nouveau mélange donne deux aires A2’ et A3’.
Démontrons maintenant le résultat suivant:
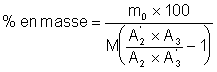
Soit M0 la masse initiale de mélange, et m0i les masses des divers constituants de ce mélange. On en pèse exactement une fraction M qui contient les masses mi des constituants. Grâce à la seringue chromatographique, on injecte dans la colonne une masse m contenant les masses mi des constituants. Nous allons nous intéresser aux constituants n°2 et n°3. Comme les rapports des masses des constituants d’un mélange se conservent dans toute fraction de ce mélange, il vient:
![]() (1)
(1)
A la masse M prépesée,
on rajoute alors une masse m0 pesée précisément de composé
n°2. En appelant m’ la masse
injectée et m’i les
masses des constituants de m’ ,
il vient: ![]() (2)
(2)

En admettant que tout ce qui a été injecté sort de la colonne, on a :
![]()
D’où nous éliminons les constantes de proportionnalité:
![]()
Le rapport de ces égalités donne:
![]() (3)
(3)
(1) et (2) nous donnent d’autre part:
![]()
En combinant (3) et (4), nous obtenons une relation où seul m2 est inconnu:
![]()
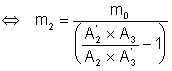
Le pourcentage en masse de 2 dans le mélange est donc:
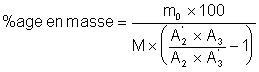
La méthode demande une bonne linéarité de la réponse du détecteur chromatographique, ce qui n’est pas toujours le cas. Nous supposerons cependant que le détecteur utilisé en TP, le catharomètre, présente cette qualité.
Étalonnage interne
Dans cette méthode, on compare individuellement chacun des pics à évaluer au pic d’une substance étalon E , convenablement choisie, introduite en proportion connue dans le mélange à analyser. il convient évidemment que le pic étalon ne soit confondu avec aucun des pics du chromatogramme.
On peut écrire: ![]() ; On
définit alors
; On
définit alors ![]()
On calculera donc la réponse de chaque soluté concerné par rapport à l’étalon. La méthode est générale. Elle est précise et reproductible.
Elle suppose néanmoins le choix d’un étalon qui, outre la nécessité de ne pas chevaucher avec les autres solutés, doit donner un pic de valeur de rétention proche de celle du pic à mesurer, d’aire approximativement égale à celle du pic du soluté, et dont la réponse doit se situer dans la zone de linéarité du détecteur utilisé.
Chromatographie:
Liste des expériences proposées
1. Chromatographie sur couche mince (CCM)
Isomérisation de l'azobenzène
Séparation des pigments du paprika
Chromatographie de parfums (Révélation par les U.V)
Chromatographie des produits résultant de l'oxydation de l'alcool benzylique (Révélation par les U.V)
Chromatographie d'acides aminés résultant de l'hydrolyse de l'aspartame (Révélation par la ninhydrine)
Chromatographie de glucides (Révélation par le réactif de Molisch)
Chromatographie de produits odorants
Etude par CCM (adsorbant: cellulose) des anthocyanines de fruits et légumes
CCM "autour de la lavande"
2. Chromatographie sur papier
Encres
Séparation de cations métalliques
Etude des colorants alimentaires des M et M's
Recherche de l'acide malique dans le vin
3. Chromatographie sur colonne
Séparation des pigments des feuilles
Séparation de colorants
Séparation des colorants d'un sirop de menthe
Détermination de la "somme des cations" d'une eau minérale en utilisant une résine échangeuse d'ions
Exemple de chromatographie d'exclusion
4. Chromatographie en phase gazeuse
Etude d'un mélange d'alcools
Méthode de l'étalon interne:
Dosage de l'éthanol du vin par CPG
Pinacol-pinacolone
5. Une chromatographie que l'on peut porter
Chromatographie sur T- shirt
CHROMATOGRAPHIE SUR COUCHE MINCE (CCM)
1. Isomérisation de l'azobenzène:
Bibliographie:
"Chimie organique expérimentale"
Auteurs:Blanchard ...Editeur : Hermann
Cette réaction peut illustrer le paragraphe sur les réactions photochimiques et le principe de la vision en option sciences expérimentales de 1ère S.
L'azobenzène f--N = N--f est commercialisé sous forme de l'isomère E. Sous l'action de la lumière, il se transforme partiellement en isomère Z.
Dissoudre 0,1 g d'azobenzène dans 4 mL de toluène et verser rapidement la solution dans un tube à essai bouché et placé dans un manchon qui le maintient à l'obscurité.
Avec un capillaire, on dépose un peu de solution à 1 cm environ du bord inférieur d'une plaque de silice pour CCM. La tache doit faire de 3 à 4 mm de diamètre. On pourra effectuer le dépôt en plusieurs fois pour éviter l'étalement. Laisser sécher et éclairer fortement pendant 1 H 30 à 2H à l'aide d'une lampe de bureau.
Déposer ensuite à 2 cm de la première tache et à même distance du bord inférieur, une tache de solution conservée à l'abri de la lumière. En éluant au toluène, on observe que la tache qui a été éclairée se dédouble: l'isomère Z formé par photoisomérisation migre plus lentement que l'isomère E, seul présent dans le produit non éclairé (l'isomère Z, plus polaire, est mieux retenu par la silice).
On pourra mesurer les Rf correspondant aux deux isomères.
2. Séparation des pigments du paprika:
Bibliographie:
"Chimie des couleurs et des odeurs"
Auteurs : Capon, Courilleau-Haverlant, Valette Editeur: Cultures et techniques, IUFM de Nantes
Placer dans un ballon de 100 mL, 2 grammes de paprika commercial et 20 mL
de dichlorométhane. Chauffer à reflux pendant 30 minutes. Filtrer. La solution
obtenue renferme tous les pigments du paprika qui ont été extraits par le
dichlorométhane.
Procéder à la CCM (3 dépôts sur la même plaque)
Adsorbant : gel de silice
Eluant : dichlorométhane
Révélateur: inutile, les pigments apparaissent sous forme de taches colorées que l'on peut facilement identifier.
Questions :
a)Quel est le nombre de pigments du paprika ?
b)Existe-t-il des pigments prépondérants ?
c) Calculer les différents Rf.
Remarques:
On trouve parfois dans un mode opératoire de CCM, le conseil suivant: passer les plaques à l'étuve pour les activer. Cette opération a pour effet de déshydrater la plaque en libérant ainsi des groupes -OH réactifs de l'adsorbant qui seront alors susceptibles de fixer les constituants polaires des mélanges à analyser.
Le paprika contient de nombreux pigments colorés qui sont facilement séparés par chromatographie. Par CCM, on obtient une large tache rouge représentant le pigment majoritaire qui donne au paprika sa couleur rouge sombre. Il est constitué d'un mélange d'esters d'acides gras de la capsanthine.
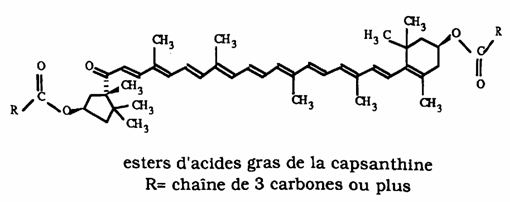
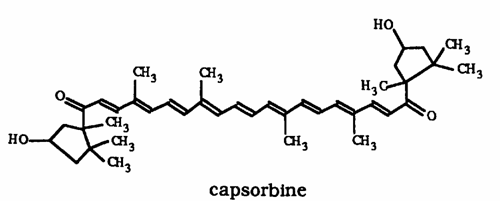
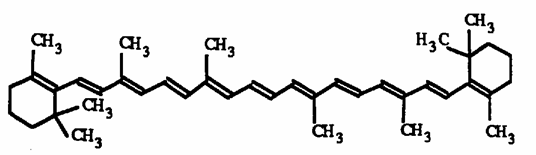
Le paprika contient d'autres caroténoïdes que la capsanthine, en particulier:
des esters d'acides gras de la capsorbine
du b- carotène:
3. Chromatographie de parfums:
Bibliographie:
"Analyse d'un parfum par chromatographie d'adsorption" BUP n° 684 p 865 à 869
Les parfums, les eaux de toilette, sont des mélanges de bases communes qui sont:
des essences naturelles (bergamote, citron,rose, cannelle, lavande)
des essences de synthèse (très diverses ,permettant d'avoir des "bouquets fleuris")
des produits odorants fournis par des animaux: ambre gris, musc, civette, castoreum
Un parfum comporte toujours:
un complexe léger fruité, s'évaporant facilement, destiné à provoquer une première senstion olfactive, passagère
le parfum proprement dit, dont l'odeur ne doit pas être fugace et qui réagit avec la peau de la personne qui le porte, ce qui le personnalise et le fait abandonner ou adopter.
Nous rechercherons dans des eaux de toilette si possible "citronnées", la présence de citral de formule (CH3)2C = CH CH2 CH2 C(CH3) = CHCHO.
Avec l'éluant utilisé, on peut mettre en évidence la présence de terpènes linéaires (C5H8)n (contenant des "unités isoprèniques" CH2 = C(CH3) CH =CH2) pour des Rf de l'ordre de 0,20 à 0,50.
Adsorbant: silice
Eluant: pour 50 mL d'hexane, 20 mL de chloroforme et 2 mL d'acétone.
Révélateur: U.V
Procéder à la CCM en déposant des taches d'eau de toilette, de citral mis en solution dans un peu d'éluant et éventuellement d'essence de citron également diluée dans un peu d'éluant.
Comparer les Rf des produits obtenus.
4. Chromatographie des produits résultant de l'oxydation de l'alcool benzylique (révélation par les U.V)
Bibliographie:
"Physique- chimie , enseignement de spécialité"
Auteurs: Durandeau, Durupthy .... Editeur : Hachette
Lors de l'oxydation de l'alcool benzylique en acide benzoïque, le produit brut obtenu, avant recristallisation, contient différentes impuretés qui sont
l'alcool de départ non oxydé ( à l'état de traces)
le benzaldéhyde résultant d'une oxydation secondaire
On se propose d'identifier ces impuretés éventuelles en réalisant une chromatographie sur
couche mince. On pourra éventuellement rechercher aussi la présence de benzaldéhyde dans l'essence d'amande amère.
Préparer des solutions à 1 % d'acide benzoïque commercial, de benzaldéhyde commercial, d'alcool benzylique commercial et du produit brut (résultant de la réaction d'oxydation de l'alcool benzylique), dans l'éther.
Procéder à la CCM des quatre solutions préparées et éventuellement de l'essence d'amande amère sur la même plaque.
Adsorbant : gel de silice sur une plaque sensible aux U.V.
Révélateur : lampe U V (cercler les taches sous la lampe)
Eluant : cyclohexane/acétone (2/1 en volume)
Exploitation:
Rappeler la définition d'un groupement chromophore.
Expliquer pourquoi les différents produits organiques étudiés apparaissent sous la forme de taches sombres lors de l'exposition aux radiations U.V..
Calculer les différents Rf.
Quelles sont les impuretés contenues dans le produit brut résultant de la réaction d'oxydation de l'alcool benzylique ?
Justifier la présence de deux taches à partir du dépôt de la solution éthérée de benzaldéhyde.
Quels corps peut-on identifier dans l'essence d'amande amère?
5. Chromatographie d'acides aminés résultant de l'hydrolyse de l'aspartame:
Bibliographie:
"Etude de l'aspartame"
Concours régional des Olympiades de chimie 91/92, centre de Strasbourg
"Additifs alimentaires et auxiliaires technologiques; chimie et santé"
Auteurs: N et M Moll Editeur: Masson
"Physique- chimie , enseignement de spécialité"
Auteurs: Durandeau, Durupthy .... Editeur : Hachette
L'aspartame (pouvoir sucrant 200 fois plus important que celui du saccharose), fut découvert par hasard en 1969 par J. Schlatter (SEARLE & CO) lors de la synthèse d'un tétrapeptide gastrique destiné à un essai biologique. Le produit intermédiaire de la réaction de synthèse, un dipeptide, constitué d'acide aspartique et de phénylalanine se révéla sucré. Searle essaya de nombreux composés de structure voisine, mais décida finalement de commercialiser le produit original qui devint connu sous le nom d'aspartame.
Les principales raisons de ce choix sont:
aucun des analogues ne pouvait être fabriqué de façon plus économique.
le pouvoir sucrant et la "qualité goût" étaient excellents
les deux constituants acide aspartique et phénylalanine sont métabolisés normalement.
Searle a spéculé sur le fait que l'aspartame avait une excellente chance de survivre aux tests les plus sévères.
Il fut commercialisé sous le nom "Nutrasweet" et a fait l'objet de dépôt de nombreux brevets. L'expiration des brevets d'exclusivité de Nutrasweet date de 1987pour le Canada; 1987-1989 pour l'Europe, 1992 pour les USA.
Stabilité:
L'aspartame est l'édulcorant de synthèse dont la stabilité a été la plus étudiée. Ce produit est relativement instable en solution et se décompose en donnant naissance à un dipeptide: aspartyl phénylalanine et à la dicétopipérazine dépourvue de pouvoir sucrant. Les étapes de la décomposition thermique de l'aspartame sont représentées ainsi:
Les deux conséquences de cette décomposition sont:
la diminution du pouvoir sucrant
l'apparition de produits secondaires dont l'éventuelle toxicité a fait l'objet d'études importantes: le méthanol qui ne devrait pas être présent à des doses toxiques, la dicétopipérazine dont les tests de toxicité se sont révélés négatifs, la phénylalanine qui, par sa présence peut provoquer des maladies chez des personnes souffrant de phénylcétonurie.
L'étiquetage des aliments et boissons édulcorées à l'aspartame doit mentionner "contient de la phénylalanine".
Les principaux paramètres qui interviennent dans la stabilité de l'aspartame sont:
le temps de stockage (diminution de l'aspartame)
l'humidité (diminution de l'aspartame)
la température (diminution de l'aspartame)
le pH a des effets variables.
La meilleure stabilité de l'aspartame en solution aqueuse à 25°C se situe entre pH 3 et pH 5, conditions réalisées dans les boissons carbonatées type colas, limonades et sodas.
D.J.A (dose journalière admissible): 0,40 mg/kg p.c.
Hydrolyse de l'aspartame:
En milieu acide, on hydrolyse les fonctions amide et ester de l'aspartame.
Placer dans un ballon 2 comprimés d'édulcorant à base d'aspartame et 20 mL d'acide chlorhydrique à 1 mol/L: les comprimés se dissolvent rapidement. Chauffer à reflux pendant 30 min.
Refroidir sous un courant d'eau froide, puis verser la solution obtenue dans dans un bécher et la neutraliser avec une solution d'hydrogénocarbonate de potassium ou de sodium à 10% jusqu'à ce que le dégagement gazeux cesse.
Chromatographie:
Porter des gants pour la manipulation pour éviter que les acides aminés de la peau se déposent sur la plaque.
Adsorbant: silice
Eluant: butan-1-ol/acide acétique/eau: 6/2/2
On dispose des solutions suivantes:
solution de phénylalanine à 1g/L
solution d'acide aspartique à 1g/l
solution de leucine à 1g/l
solution de lysine à 1g/l
solution d'aspartame hydrolysé
solution fraîche d'aspartame obtenue par dissolution d'un comprimé de 20 mg dans 20 mL d'eau.
A l'aide de capillaires (un par solution), disposer une goutte de solutions de phénylalanine, d'acide aspartique, d'aspartame hydrolysé, d'aspartame frais et éventuellement de leucine et de lysine si l'édulcorant en contient et si la taille de la plaque le permet.
Eluer.
Sécher la plaque, pulvériser une solution à 2% environ de ninhydrine dans l'acétone, sous la hotte. Chauffer pour faire apparaître les taches colorées et identifier les acides aminés présents dans les solutions fraîche et hydrolysée d'aspartame.
Les réactions entre la ninhydrine et les acides aminés sont détaillées dans l'annexe (II).
6. Chromatographie de glucides:
Bibliographie:
BUP n° 774 mai 1995 p 960-961
"Deuxième recueil d'épreuves sélectionnées des Olympiades Nationales de la Chimie" p93 à 95
"Physique- chimie, enseignement de spécialité"
Auteurs: Durandeau, Durupthy... Editeur: Hachette
Préparer 100 mL de solutions à 2,5 g.L-1 de fructose, glucose et saccharose.
Placer 10 mL d'une ampoule de jus de fruit pour bébé dans une fiole jaugée de 50 mL et compléter au trait de jauge avec de l'eau déminéralisée.
Prélever 10 mL de cette solution et les placer dans un ballon. Ajouter 5 mL d'acide chlorhydrique à 2 mol.L-1 et chauffer pendant 20 min à reflux léger.
Eluant: butan-1-ol/acétone/eau: 5/4/1
Faire un dépôt des solutions de glucose, fructose, saccharose, jus de fruits dilué, jus de fruit hydrolysé.
Eluer .
Sécher la plaque.
Révéler la plaque: les taches de sucres n'étant pas visibles, on utilisera un révélateur, le réactif de Molisch qui forme des dérivés colorés avec les glucides.
Pour cela on pulvérisera du réactif de Molisch sur les plaques (hotte; gants) et on placera la plaque sur une plaque chauffante ou dans une étuve jusqu'à obtention de taches colorées.
Mesurer les Rf. Identifier les glucides présents dans le jus de fruit et dans le jus de fruit hydrolysé.
Le réactif de Molisch est préparé ainsi:
0,50 g d'a- naphtol dans 100 mL d'éthanol + 100 mL d'acide sulfurique à 20%
Les réactions mises en jeu sont évoquées dans l'annexe (III).
7. Chromatographie de produits odorants:
Bibliographie:
"Journal of Chemical Education" decembre 95 page 1137 à 1138
On peut analyser par chromatographie sur couche mince des produits odorants purs et des mélanges quelconques de ces produits.
On dispose de:
citral ou 3,7-diméthylocta-2,6 diénal, Z et E:
citronellol ou 3,7-diméthyl oct-6-ènol:
eugénol ou4-allyl 2-méthoxyphénol:
limonène:
linalol ou 3,7-diméthyloct-2,6-diène-1-ol
menthol
On utilise des plaques de silice.
L'éluant est un mélange toluène/acétate d'éthyle (proportions 90/10 en volume).
Les solutions à analyser sont préparées ainsi:
3 gouttes (si produit est liquide) dans 5 mL de dichlorométhane
0,05 g (si le produit est solide) dans 5 mL de dichlorométhane
On pourra réaliser des mélanges également.
On préparera deux plaques de CCM identiques car on dispose de deux révélateurs:
solution de permanganate de potassium (dissoudre 1,6 g de permangante de potassium et 15 g de carbonate de sodium anhydre dans 100 mL d'eau déminéralisée; vérifier qu'il n'y a pas formation de dioxyde de manganèse brun)
solution de DNPH (dissoudre 0,1 g de 2,4-dinitrophénylhydrazine dans 100 mL d'éthanol à 95% et 1 mL d'acide chlorhydrique à 12 mol/L)
Lorque l'élution est terminée, vaporiser sur une plaque la solution de permanganate de potassium et sur l'autre la solution de DNPH.
La solution de permanganate de potassium donne des taches brunes (MnO2) avec tous les composés odorants (à vérifier avec le menthol), tandis que la solution de DNPH ne réagit qu'avec les composés carbonylés (citral).
L'article du Journal of Chemical Education donne les valeurs des Rf pour de nombreux produits odorants:
citronellal (0,72)- piperonal (0,49)- menthol (0,26)- citronellol (0,20)- 4-allylanisole (0,66)- acétate de linalyle (0,55)- carvone (0,45) - cuminaldéhyde (0,57)- eugénol (0,44)- vanilline (0,19) - citral (0,48)- linalol (0,33) - limonène (0,72) - anisaldéhyde (0,44).
8. Etude par CCM (adsorbant: cellulose) des anthocyanines de fruits et légumes:
Bibliographie:
"Journal of Chemical Education" Avril 96 page 306 à 309
"Chimie des couleurs et des odeurs" Association Cultures et Techniques Nantes
Principe:
Les couleurs rose, mauve, violette et bleues des pétales de fruits et des légumes sont dues à la présence d'anthocyanines, qui sont des flavonoïdes c'est à dire des molécules dérivant du noyau flavone
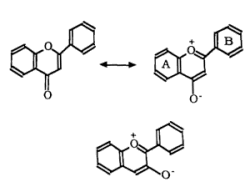
Ce sont des hérérosides substitués du cation flavylium:
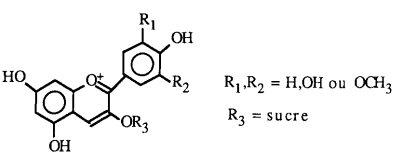
Selon la nature du sucre R3, on obtient une grande variété d'anthocyanines. Par hydrolyse acide, on peut casser la liason avec le sucre et obtenir des aglycones appelés anthocyanidines. Il n'existe que 6 anthocyanidines existant à l'état naturel:
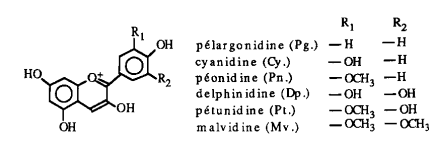
Voici la liste de quelles anthocyanidines présentes dans quelques fruits et légumes:
|
Fruit ou légume |
pomme |
myrtille |
airelle |
chou rouge |
fraise |
|
Anthocyanidine |
Cy. |
Mv. Pt. Dp. |
Cy. Pn. Dp. |
Cy. |
Pg. Cy. |
Par chromatographie sur couche mince, on pourra mettre en évidence le nombre d'anthocyanines présentes dans le fruit et le légume et éventuellement les anthocyanidines obtenues après hydrolyse acide. les composés étant colorés, il ne sera pas nécessaire d'utiliser un révélateur pour les plaques.
Expérience:
Extraction des anthocyanines:
On extrait les anthocyanines en couvrant 5 à 10 g de fruit ou de légume (ou de peau de pomme rouge) d'un mélange méthanol/acide chlorhydrique concentré (proportions 99/1 en volume). L'extraction nécessite environ une heure. Si l'extrait doit subir ensuite une hydrolyse, on pourra le concentrer en utilisant l'évaporateur rotatif ou en laissant le récipient ouvert sous une hotte.
Hydrolyse des anthocyanines:
Mélanger volume à volume , la solution contenant les pigments précédemment extraits et de l'acide chlorhydrique à 4 mol/L dans un tube à essais que vous munirez d'un réfrigérant à air. Chauffer au bain-marie à 80°C pendant une durée variable (il faut environ 30 minutes pour une hydrolyse partielle et 1 heure pour une hydrolyse totale dans le cas de pomme ou des autres fruits mais plus longtemps pour le chou rouge qui est un diglycoside alors que les autres sont des monoglycosides.
Chromatographie sur couche mince:
On utilisera une plaque dont la phase stationnaire est de la cellulose. Il faudra faire plusieurs dépôts pour obtenir une tache de petite taille mais bien colorée.
L'éluant est constitué d'un mélange acide chlorhydrique concentré/ acide méthanoïque/eau dans les proportions volumiques 19,0/39,6/41,4.
On observera une ligne de l'éluant moins nette qu'en chromatographie sur couche mince avec support silice.
Résultats:
On peut mettre enévidence le nombre d'anthocyanines présentes dans les différents extraits avant hydrolyse et mesurer les Rf correspondant.
L'article du Journal of Chemical Education donne : Pomme (0,47)- Myrtille (0,32 - 0,47 - 0,60)- Fraise (0,44- 0,60) - Airelle (0,49- 0,62) - Chou rouge (0,93).
Les anthocyanidines obtenues après hydrolyse sont moins polaires et migrent moins sur la plaque. On peut mettre en évidence après hydrolyse, la présence des mêmes anthocyanidines dans des solutions provenant de fruits différents
9. CCM "autour de la lavande":
Bibliographie:
Préparation aux Olympiades de chimie (Besançon 95/96 et Nancy-Metz 96/97)
"Aspic, lavande et lavandin" BUP n° 789 décembre 96 p 1941 à 1950
On peut:
extraire de l'huile essentielle de lavande
synthétiser de l'éthanoate de linalyle, constituant présent dans la lavande et dans la bergamote
comparer par CCM: l'éthanoate de linalyle, l'huile essentielle de lavande, le linalol, l'essence de bergamote, un parfum "lavande" (on dispose de ces produits)
Extraction de l'huile essentielle de lavande:
On adaptera le mode opératoire au matériel présent.
Réduire de la lavande en poudre fine (mortier ou moulin à café) pour obtenir environ 200 mL de poudre fine
Mettre la poudre dans un ballon de 1 litre contenant environ 600 mL d'eau et réaliser une hydrodistillation
On obtient une fine pellicule d'huile essentielle: on ajoutera quelques spatules de chlorure de sodium pour faciliter la séparation de l'essence.
Introduire le distillat dans une ampoule à décanter, agiter, laisser reposer. Après séparation des 2 phases, évacuer la phase aqueuse puis recueillir l'huile essentielle qui surnage.
Si on obtient suffisamment d'huile essentielle, on pourra ajouter une pointe de spatule de sulfate de sodium pour déshydrater l'huile essentielle.
Si la quantité d'huile essentielle obtenue par hydrodistillation est trop faible, on pourra la récupérer par extraction à l'éther et élimination ensuite de l'éther à l'évaporateur rotatif ou avec deux fioles à vide (une de garde).
Synthèse de l'éthanoate de linalyle:
L'éthanoate de linalyle est un ester présent dans la lavande (de 35 à 55%) et dans la bergamote (de 35 à 45 %). Il peut être obtenu par action de l'anhydride éthanoïque sur un alcool tertiaire possédant des doubles liaisons: le 3,7- diméthyloct-1,6-diène-3-ol ou linalol.
Dans la lavande on trouve également de l'éthanoate de géranyle, qui peut être obtenu par action de l'anhydride éthanoïque sur le géraniol ou 3,7-diméthyloct-2,6-diène-1-ol.
Introduire dans un ballon 10 mL de linalol et 20 mL d'anhydride éthanoïque (sans rincer l'éprouvette graduée)
Adapter sur le ballon le réfrigérant à reflux.
Mettre en route la circulation d'eau froide.
Chauffer à l'aide d'un chauffe-ballon pendant 30 min à partir de l'ébullition.
Arrêter le chaffage, introduire par le sommet du réfrigérant 25 mL d'eau déminéralisée.
Laisser refroidir le contenir du ballon , éventuellemnt en refroidissant sous un filet d'eau.
Tranvaser dans l'ampoule à décanter
Eliminer la phase aqueuse
Verser dans l'ampoule à décanter 20 mL de solution d'hydrogénocarbonate de sodium à 5%. Agiter. Attention au dégagement gazeux.
Recommencer la même opération avec 20 mL d'eau déminéralisée.
Placer l'ester dans un erlen, ajouter environ 2 g de K2CO3 anhydre. Boucher l'erlen et agiter quelques instants pour sécher la phase organique
Recueillir le liquide surnageant.
|
|
linalol |
anhydride acétique |
éthanoate de linalyle |
|
Densité |
0,87 |
1,08 |
0,89 |
|
Température d'ébullition |
199 |
139,5 |
220 |
|
Solubilité dans l'eau |
non |
oui |
non |
Caractérisation par C.C.M:
On dispose:
d'éthanoate de linalyle préparé
d'huile essentielle de lavande
d'un parfum "lavande"
d'essence de bergamote
de linalol
Préparer un mélange cyclohexane/ éthanoate d'éthyle (proportions volumiques (8/2)) qui servira à la fois d'éluant et de solvant.
Préparer la cuve chromatographique.
Préparer les solutions à analyser en mettant dans un tube à essais 3 mL du mélange cyclohexane/ éthanoate d'éthyle et une goutte du produit étudié. Procéder de la même façon pour les cinq produits.
Procéder à la C.CM.
Révélation: I2 (+ sable) dans un flacon bouché.
Variante:
L'article du BUP propose de diluer les produits à étudier à raison de 1 goutte dans 1 mL de dichlorométhane et d'utiliser le dichlorométhane comme éluant.
CHROMATOGRAPHIE SUR PAPIER
1. Chromatographie d'encres de feutres:
Découper une bande de papier Whatman n° 1, suffisamment large pour déposer toutes les taches de feutres. La bande sera disposée en cylindre.
Eluant: butan-1-ol/ éthanol/ammoniac à 2mol.L-1: 6/2/2
Réaliser la chromatographie.
Application: identifier les colorants purs et les mélanges.
2. Séparation de cations métalliques:
Bibliographie:
Journal of Chemical Education, Avril 93
Eluant: acétone/ acide chlorhydrique à 6 mol.L-1: 4/1
On dispose de solutions de nitrate, sulfate ou chlorure de nickel, cobalt, cuivre à environ 5.10-2mol/L.
Faire, à l'aide de capillaires, des taches de chacune de ces solutions et une tache d'un mélange de ces solutions à environ 1 cm du bord inférieur de la bande de papier Whatman n° 1.
Placer le papier sous forme d'un demi-cylindre et éluer (30 à 40 min).
Sécher le papier à l'air libre pendant 10 min avant de le placer dans une chambre à ammoniac ( 5 mL d'ammoniac concentré dans un bécher de 30 mL, lui-même placé dans un bécher sec de 600 mL) sous la hotte.
Quand la couleur bleue caractéristique de Cu(II) apparaît, enlever la papier de la chambre à ammoniac et appliquer avec soin une goutte de diméthylglyoxime à l'aide d'un compte-goutte sur la tache initiale de la solution de nickel et sur la tache initiale du mélange. Une couleur rose indique la présence de Ni(II) .
Placer le papier dans une chambre à sulfure d'ammonium (préparation identique à celle à ammoniac). Les cations donnent des précipités de couleur brune à noire.
Valeurs indiquées dans l'article pour les Rf:
Ni2+: 0,08 Co2+: 0,35 Cu2+: 0,60
3. Etude des colorants alimentaires des M & M's:
Bibliographie:
" Séparation de colorants par chromatographie" BUP n° 750 p 59
" Chromatography of M& M's candies" Journal of Chemical Education Dec 92
" Additifs alimentaires et auxiliaires tecnologiques"
Auteurs: N et M Moll Editeur: Masson
Manipulation:
Les colorants alimentaires sont des addtifs alimentaires qui ont un numéro de code C.E.E de 100 à 199 précédé de la lettre E.
Cette manipulation peut être effectuée en option sciences expérimentales en 1ère S.
On dispose tout d'abord d'un lot de trois colorants alimentaires commercialisés sous la marque Vahiné:
colorant jaune:E 102, tartrazine
colorant rouge: E 122, azorubine
colorant vert: mélange de tartrazine (Jaune, E 102) et de Bleu Patenté V (Bleu E 131)
D'autre part, on dispose de bonbons chocolatés "M & M's" contenant les colorants
E 104, E 110, E 122, E 124, E 131, E 171
Les formules de ces colorants, leurs utilisation ainsi que la D.J.A (dose journalière admissible exprimée en mg/kg de poids corporel mg/kg p.c) figurent dans l'annexe (IV) .
Vous pourrez réaliser sur une même bande de papier Whatman n° 1, la chromatographie sur papier de chacun des colorants Vahiné, d'un colorant M & M's et ensuite à exploiter ce chromatogramme.
On découpera une bande de papier Whatman n° 1 de hauteur telle qu'elle tienne comme indiqué dans le bocal.(en évitant de mettre ses doigts sur le papier !!)
A 2 cm du bord inférieur du papier Whatman, on placera à un centimètre l'une de l'autre quatre taches ainsi, en commençant à environ 1 cm du bord:
une tache de chaque colorant Vahiné. Pour chaque tache, on prendra un capillaire différent.
à l'aide d'un cure-dent que l'on mouillera, on grattera le bonbon de la couleur choisie pour extraire le colorant. Il faut faire attention à ne pas atteindre le chocolat. Le cure-dent est essuyé doucement sur du papier filtre ou sur de l'essuie-tout et l'extrait est appliqué d'un mouvement rapide à sa place prévue sur le papier Whatman. On pourra faire 2 applications successives au même endroit pour avoir une tache bien colorée.
Sécher la bande de papier Whatman au sèche-cheveux , puis placez-la avec la pince à dessin dans un bocal où vous aurez versé auparavant 1 cm de haut environ de solution de chlorure de sodium à 0,1% en masse (éluant) ;l'éluant ne doit pas atteindre la ligne de dépôt en début d'expérience.
Observez la migration pendant 10 à 20 minutes: le solvant doit arriver à 2 cm du haut du papier Whatman environ.
Noter rapidement au crayon de papier les positions du solvant (front du solvant) et de chaque tache. Séchez la feuille de papier Whatman rapidement au sèche-cheveux.
Mesurer les Rf de chaque constituant.Pour les taches étalées, on prendra le milieu de la tache.
Exploitation:
Montrer que le colorant utilisé sur les M & M's jaunes n'est pas de tartrazine (suspectée allergisante)
Rechercher la présence éventuelle des colorants E 122 et E 131 dans les colorants alimentaires des M & M's.
Identifier le ou les colorants présents dans l'enrobage d'un M& M's à l'aide du chromatogramme et des données figurant dans l'annexe (IV).
Savoir si l'enrobage d'un M&M's de couleur donnée est un colorant pur ou un mélange
4. Recherche de l'acide malique dans le vin:
Bibliographie:
"Recherche et évaluation de l'acide malique par chromatographie"
BUP n° 775, juin 95 p 1181
" Séparation des acides d'un vin par CCM"
Deuxième recueil d'épreuves sélectionnées des Olympiades Nationales de la Chimie
La préparation d'un vin à partir de jus de raisin commence par la fermentation alcoolique qui conduit à la transformation des sucres en alcool. On peut arrêter l'évolution à ce stade par filtration. C'est le cas des vins blancs.
Le produit obtenu contient alors, à l'état de traces, plusieurs composés acides:
acide malique: HOOC-CH2-CHOH-COOH
acide tartrique HOOC-CHOH-CHOH-COOH
acide succinique HOOC-CH2-CH2- COOH
acide lactique HOOC- CHOH-CH3
L'acide malique du vin est biologiquement instable. Sa teneur diminue au cours de la fermentation alcoolique et devient nulle après la fermentation malolactique (deuxième fermentation au cours de laquelle l'acide malique se transforme en acide lactique); cette fermentation malolactique est recommandée pour la plupart des vins rouges car cela diminue leur acidité.
Les vins peuvent contenir jusqu'à 5g/L d'acide malique.
Une méthode simple par chromatographie sur papier permet de savoir si le vin contient ou non de l'acide malique et d'en apprécier la teneur. Cette chromatographie est surtout réalisée pour suivre l'évolution de la fermentation malolactique.
Eluant (à préparer au moment de l'emploi):
40 mL de solution A de butan-1-ol à 1 g/L de bleu de bromophénol
20 mL de solution B d'acide éthanoïque à 50%
Vous disposez de:
vin blanc
vin rouge
solution d'acide malique à 10g/L
solution d'acide tartrique à 10 g/L
solution d'acide lactique à 10 g/L
solution d'acide succinique à 10 g/L
papier Whatman n° 1
Déposer en des positions préalablement repérées à 1 ou 2 cm du bord inférieur de la feuille de papier Whatman, à l'aide de capillaires, des taches des solutions à analyser.
On obtiendra des taches plus intenses et moins étalées en renouvelant les dépôts aux mêmes endroits et en séchant la feuille entre chaque opération au sèche-cheveux.
Après élution, on marque le front du solvant puis on fait sécher la feuille sous une hotte bien ventilée: le papier initialement imbibé de solvant acide (BBP jaune) , devient bleu-violet sauf aux endroits où se situent les acides du vin.
Après révélation des taches du vin, on peut observer trois spots d'acides:
acide tartrique à la partie inférieure
acides lactique et succinique non séparés à la partie supérieure
éventuellement acide malique au centre
CHROMATOGRAPHIE SUR COLONNE
1. Séparation des pigments des feuilles:
Bibliographie:
Journal of Chemical Education Décembre 92 p 987 et 988
Cette expérience peut être réalisée à partir de feuilles d'arbres, de buissons, de plantes d'intérieur, de feuilles de salades colorées....
Deux petites feuilles sont écrasées dans un mortier en présence de 2 à 3 mL d'acétone pour réaliser un extrait. Le liquide est extrait à l'aide d'un compte-goutte pour le séparer de la pulpe.
La colonne est contituée d'hydrogénocarbonate de sodium dans l'hexane ou dans l'éther de pétrole (préparation par voie sèche ou par voie humide) sur 2 ou 3 cm de haut.
Le niveau de l'éluant est placé au niveau supérieur de l'hydrogénocarbonate de sodium et l'extrait "jus de feuilles" est placé avec précaution en haut de la colonne. On ajuste à nouveau le niveau au ras de l'hydrogénocarbonate de sodium.
On élue tout d'abord à l'éther de pétrole: une bande d'un vert lumineux se sépare: elle est composée essentiellement de chlorophylle et de pigments caroténoïdes; elle peut être recueillie dans un premier tube à essais.
Quand cette première bande est éliminée de la colonne, on change d'éluant: on utilise alors de l'acétone. Selon les espèces, la bande éluée a une coloration vert pâle à jaune due à des pigments phytochromes.
Quand la bande éluée à l'acétone a été récupérée, l'éluant est à nouveau changé pour un mélange propan-2-ol/eau en proportions volumiques 7/3. Deux ou trois bandes de coloration jaune-brune apparaissent.
Une dernière bande de coloration rouge-brun est enfin éluée par une solution aqueuse saturée en hydrogénocarbonate de sodium.
Ces dernières bandes, éluées par le mélange propan-2-ol/eau et par la solution d'hydrogénocarbonate de sodium contiennent des pigments flavonoïdes solubles dans l'eau.
2. Séparation de colorants:
Bibliographie:
Journal of Chemical Education Décembre 92 p 991 et 992
Attention, les colorants utilisés marquent les tissus. Utilisez des gants!!
Un mélange de 100 mg de rhodamine B base et de 100 mg de diphénylthiocarbazone ou dithizone dissous dans 10 mL d'acétone a été préparé peu avant ce stage car le colorant vert (diphénylthiocarbazone) s'estompe à la longue.
Expérience préalable:
Placez dans un bécher 10 mL d'eau et 10 mL d'heptane puis une goutte du mélange de colorants. Agitez si nécessaire pour accélérer la séparation: on observe une phase inférieure (phase aqueuse) rose brillant (coloration due à la rhodamine B base) et une phase supérieure (heptane) verte (coloration due à la dithizone).
Elution du colorant vert sur colonne:
Préparation de la colonne:
Mettre au fond d'une pipette Pasteur, un peu de coton puis la remplir au 2/3 de gel de silice et ajouter un peu de sable en haut. Humidifier avec environ 5 gouttes d'heptane.
Elution:
Ajouter 2 gouttes du mélange de colorants en haut de la colonne de silice, au centre.
Laisser le mélange migrer jusqu'à ce qu'il atteigne le haut de la colonne.
Ajouter de l'heptane pour remplir la pipette. Eluer le colorant vert en renouvelant les ajouts d'heptane si nécessaire et veillant à ce que la colonne ne s'assèche pas.
On sépare ainsi le pigment vert du pigment rouge qui reste lui sur la colonne.
3. Séparation des colorants d'un sirop de menthe:
Bibliographie:
" Séparation des colorants d'un sirop de menthe par chromatographie"
BUP n° 756 Juillet-août-septembre 93
Préparation de la colonne:
Placer un petit morceau de coton au fond d'une pipette de Pasteur. Verser une spatule de silice dans de l'eau. Mettre en suspension, puis verser dans la pipette Pasteur maintenue verticale.
Elution:
Dès que le niveau d'eau atteint le lit de la colonne, déposer à l'aide d'un compte-gouttes deux gouttes de sirop de menthe, puis maintenir immédiatement la colonne pleine d'eau à ras bord.
Le premier colorant est élué à l'eau: il s'agit de la tartrazine (voir annexe IV sur les colorants alimentaires). On peut tracer rapidement son spectre à l'aide du spectrophotomètre placé dans la salle.
Eluer le second colorant à l'aide d'éthanol à 95%: il s'agit de bleu Patenté V; on pourra également tracer son spectre.
On peut comparer ces spectres avec les spectres des colorants Vahiné:
colorant jaune (tartrazine)
colorant vert (mélange de tartrazine et de bleu patenté V)
On pourra également tracer le spectre d'une solution aqueuse de sirop de menthe.
Variante :
On peut également extraire les colorants du sirop de menthe en les fixant sur de la laine puis les extraire de celle-ci en utilisant le fait que la laine ne fixe ces colorants anioniques qu’en milieu acide.
On place dans un bécher 4 à 5 brins de laine incolore de 20 cm puis environ 40 mL de sirop de menthe et 5 mL d’une solution d’acide acétique à 2 mol/L. On porte le mélange à ébullition douce pendant 10 minutes : la laine prend une couleur verte intense alors que la solution devient presque incolore. A l’aide d’un agitateur, on retire la laine et on la rince à l’eau (bonne tenue de la teinture). On introduit ensuite cette laine teintée dans un bécher contenant environ 15 mL d’une solution d’ammoniac à 0,1 mol/L et on porte à ébullition douce. Rapidement la solution prend une couleur verte. On retire la laine et on laisse bouillir la solution quelques instants afin de la concentrer par évaporation de l’eau. On utilisera cette solution pour faire une C.C.M (plaque de silice) en la comparant à des solutions de tartrazine et éventuellement de bleu patenté. L’éluant sera constitué d’un mélange éthanol, solution de chlorure de sodium à 40 g/l dans les proportions volumiques 1/5.
On peut faire de même avec un sirop de grenadine contenant de l’azorubine (E 122) et du rouge cochenille (E 124) ou un sirop d’orange contenant du jaune orangé S (E 110).
4. Détermination de la "somme des cations" d'une eau minérale en utilisant une résine échangeuse d'ions:
Généralités sur les résines échangeuses d'ions (voir aussi annexe V)
Les échangeurs d'ions sont des solides insolubles qui ont la propriété de pouvoir échanger leurs ions avec ceux d'une solution. Ce sont en général des résines synthétiques constituées par un réseau macromoléculaire (le plus souvent du polystyrène) sur lequel sont greffés des radicaux ionisables ou ionisés, dites soit cationiques lorsque elles peuvent échanger leurs cations (tels que H+), soit anioniques lorsqu'elles peuvent échanger leurs anions (tels que OH- ou Cl-).
Les résines sont généralement utilisées en colonnes„ en faisant couler très doucernent la solution dans la colonne. Lorsque l'on veut séparer les constituants d'un mélange, on choisit les conditions opératoires de telle sorte qu'un des constituants du mélange ne soit pas retenu sur la résine, et que l'autre constituant soit fixé sur la résine. On modifie ensuite les conditions opératoires en faisant couler sur la résine un solvant approprié, appelé éluant, tel que le constituant fixé sur la resine soit entraîne par l'éluant. Cette technique s'appelle la chromatographie par échange d'ions.
Précaution opératoire : la résine ne doit jamais être en contact avec l'air : elle doit toujours être surmontée d'un peu de liquide.
Principe de la manipulation:
L'eau minérale à analyser (Hépar ou Contrexéville) contient comme cations Ca2+, Mg2+, Na+ et K+. L'eau à analyser est passée sur une résine cationique(IR 120) .
Les cations Ca2+,Mg2+,K+,Na+ sont remplacés par des ions ions H+ fournis par une résine échangeuse d'ions fortement acide selon la réaction:
Mn+(solution) + nH+(résine) Mn+(résine) + nH+(solution)
L'éluat contient donc comme espèces ayant des propriétés acidobasiques des ions H3O+ provenant du passage sur la résine et des ions HCO3- provenant de l'eau minérale. On portera l'éluat à ébullition pendant quelques minutes de telle sorte que la réaction suivante ait lieu:
H3O+ + HCO3- CO2 + 2 H2O
Après refroidissement, on dosera par pH-métrie l'éluant à l'aide d'une solution d'hydroxyde de sodium de concentration connue, voisine de 5,00 . 10-2 mol/L.
Mode opératoire:
Verser à l'aide d'une éprouvette graduée 200 ml d'eau déminéralisée dans un bécher et 50 ml d'acide chlorhydrique au demi.
Faire passer lentement cette solution sur la colonne de résine pour la convertir en résine acide.
Veiller à ce que la colonne de liquide ne descende jamais au-dessous du sommet du lit de résine.
Rincer la colonne avec de l'eau déminéralisée jusqu'à élimination totale des chlorures.
Contrôler cette élimination par l'absence de précipité avec une solution de nitrate d'argent .
Faire passer lentement sur la résine E=20 ml d'eau minérale.
Recueillir l'éluat dans un bécher de 250 ml.
Rincer 3 fois la colonne avec 20 ml d'eau déminéralisée et recueillir les eaux de lavage dans le même bécher.
Porter à ébullition pendant 5 min pour chasser le dioxyde de carbone. Laisser refroidir.
Titrer par pH-métrie à l'aide de la solution de soude étalonnée.(Soit Ve le volume équivalent.)
Vérifier la relation donnant la "somme des cations" à partir des indications fournies par le fabricant sur l'étiquette de l'eau minérale:
2 [Ca2+] + 2 [Mg2+] + [Na+] + [K+] - [HCO3-] =
Remarque:
On peut trouver une expérience utilisant une résine échangeuse d'ions analogue dans un article publié dans le n° 762 du BUP (Mars 1994) et intitulé:" Dosage du magnésium dans un médicament"
5. Exemple de chromatographie d'exclusion:
Bibliographie:
Journal of Chemical Education Décembre 92 p 993 et 994
Préparation du gel Sephadex:
Le Sephadex est un gel fait à partir de polyosides bactériens: les dextrans. Les Sephadex présentent une grande affinité pour l'eau, ils s'hydratent fortement, fixant jusqu'à dix fois leur masse d'eau, et gonflent jusqu'à former un réseau dont le nombre et les dimensions des mailles sont déterminés par les liaisons établies dans l'édifice moléculaire formé par le dextran hydraté.
Les gels polyosides sont insolubles dans l'eau et dans les solutions salines, stables dans les solutions alcalines ou faiblement acides mais hydrolysés par les acides forts. de plus ces gels doivent être protégés d'une hydrolyse enzymatique consécutive à une contamination bactérienne. Pour cela le gel hydraté sera stocké dans une solution de chlorure de sodium saturée.
Le gel a été préparé en ajoutant 1 g de Sephadex à environ 20 mL d'eau et l'ensemble a gonflé pendant 1 heure: on peut ainsi réaliser 10 à 20 colonnes.
Au fond de la colonne formée d'une pipette Pasteur, on placera un petit morceau de papier filtre; ensuite on ajoutera à l'aide d'un compte-gouttes, les "perles" de Sephadex hydraté jusqu'à ce que la colonne soit remplie à environ 1/2 à 2/3.
A aucun moment, la colonne ne doit être à sec!!
Elution de la solution de diiode:
On dispose d'une solution de diiode dans un compte-gouttes préparée à raison de 250 mg de I2 dans 100 mL d'éthanol à 50%.
Dès que la colonne est prête, ajouter une goutte de solution de diiode en haut de la colonne. Laver constamment à l'eau (pas plus de quelques gouttes en haut de la colonne).
Récupérer les gouttes dans un tube à essais ou un petit bécher qui contient quelques gouttes d'une solution aqueuse saturée d'amidon..
Compter le nombre de gouttes qui tombent à partir du moment où on place la solution de diiode en haut de la colonne et jusqu'à ce que la coloration de la solution du tube collecteur change.
Elution de la solution d'amidon:
Placer une goutte de solution d'amidon en haut de la colonne et éluer à l'eau en recueillant l'éluat dans un tube contenant quelques gouttes de la solution de diiode. Compter de même le nombre de gouttes qui tombent à partir du moment où on place la solution d'amidon en haut de la colonne et jusqu'à ce que la coloration de la solution du tube collecteur change.
Elution de la solution de diiode, suivie par la solution d'amidon:
Placer une goutte de solution de diiode en haut de la colonne. Eluer à l'eau et récupérer l'éluat dans un tube vide. Quand la bande jaune-brune du diiode est à peu près à mi-hauteur dans la colonne,ajouter une goutte de solution d'amidon en haut de la colonne et continuer à éluer à l'eau. Noter les éventuels changements de couleurs. Eluer le diiode en totalité.
Séparation diiode/amidon
Mélanger 2 gouttes de solution de diiode et deux gouttes de solution d'amidon. Ajouter une goutte de ce mélange en haut de la colonne et éluer à l'eau en récupérant l'éluant dans un tube vide.Compter de même le nombre de gouttes qui tombent à partir du moment où on place le mélange en haut de la colonne et jusqu'à ce que la coloration de l'éluat change. Noter les éventuels changements de couleurs dans la colonne.
CHROMATOGRAPHIE EN PHASE GAZEUSE
1. Etude d'un mélange d'alcools
Vous trouverez ci-joints (Annexe VI) les chromatogrammes de trois alcools purs et celui d'un mélange de ces trois substances.
Les valeurs lues au niveau des pics correspondent aux temps de rétention.Les manipulations ont été réalisées sur une colonne dont la phase stationnaire est polaire (carbowax). Les différents paramètres sont :
température de l'injecteur : 220°C
température du détecteur:220°C
température du four:85°C
température du filament:285°C
pression d'hélium:6 bar
atténuation:128.
Y-a-t-il une relation entre les temps de rétention et les points d'ébullition ?Justifier.
Faire l'analyse chromatographique d'un mélange inconnu de deux de ces alcools.(un certain nombre de mélanges sont déjà préparés dans des piluliers )
Injecter 1 µL
Déduire du chromatogramme obtenu
- la composition qualitative du mélange
- la composition quantitative du mélange
Attention : il est important de bien rincer à l'acétone (plusieurs fois) puis avec le produit à injecter et de bien essuyer la seringue avant chaque nouvelle injection.
2. Méthode de l'étalon interne
Exemple n °1: Dosage de l'éthanol du vin par CPG
Bibliographie:
"Chimie tout, expériences commentées" Auteurs:Haurat-Bentolila; Lecorgne; Leduc
Editeur: Association Cultures et Techniques- Nantes
Principe:
On utilisera ici la méthode de l'étalon interne.
On préparera une série de mélanges contenant, pour 10 mL de solution, des pourcentages en volume d'éthanol variant de 3 à 21%. A chaque mélange, on ajoutera toujours le même volume de propan-1-ol (1 mL).
On injectera ensuite 0,3 µL de chacune des solutions ainsi réalisées. En réalité, on n'est pas toujours certain d'injecter exactement le volume de 0,3 µL (présence de bulles dans la seringue..). Cependant comme la concentration en propan-1-ol est la même dans toutes les solutions, le mesure du rapport : aire du pic éthanol/ aire du pic propanol est proportionnelle à la concentration en éthanol dans le mélange et ne dépend pas du volume versé. On pourra donc tracer une courbe d'étalonnage et l'utiliser pour déterminer la teneur en éthanol dans le vin.
Manipulation:
Préparer dans des piluliers, les mélanges suivants:
|
% éthanol (v/v) |
3 |
6 |
9 |
12 |
15 |
18 |
21 |
|
éthanol (mL) |
0,3 |
0,6 |
0,9 |
1,2 |
1,5 |
1,8 |
2,1 |
|
eau déminéralisée (mL) |
9,7 |
9,4 |
9,1 |
8,8 |
8,5 |
8,2 |
7,9 |
Ajouter dans chaque pilulier, 1,0 mL de propan-1-ol.
Préparer de même un pilulier contenant 10,0 mL de vin et 1,0 mL de propan-1-ol.
Injecter les différentes solutions avec les réglages suivants:
température du four: 45°C
température du détecteur et de l'injecteur: 200 °C
pression d'hélium: 6 bars
température du filament réglée à 285°C pour avoir i= 200 mA dans le catharomètre
Relever les aires correspondant aux pics de l'éthanol et du propan-1-ol.
On donne les températures d'ébullition sous un bar:
propan-1-ol: 97°C éthanol: 78,3°C
Tracer la courbe aire du pic éthanol/ aire du pic propan-1-ol en fonction du % en éthanol.
En déduire le pourcentage en éthanol du vin.
Vous trouverez en Annexe VII, un exemple de résultats expérimentaux.
Exemple 2 : pinacol - pinacolone
Lorsqu'on traite le pinacol (2,3-diméthylbutane-2,3-diol) par de l'acide bromhydrique à 48 % et à chaud, il se forme :
de la pinacolone (2,2-diméthybutan-3-one) résultant d'une transposition ou réarrangement pinacolique.
du 2,3-diméthylbuta-1,3-diène résultant de la déshydratation "normale" du diol
Afin d'étudier correctement la composition du mélange en CPG, il nous faut envisager la méthode dite de l'étalon interne. On choisit pour cela la méthylisobutylcétone ou 4-méthylpentan-2-one.
Manipulation.
Dans des piluliers, préparer 2 mL de solution Ai(i = 1 , 2 ou 3)à 25 % , 50 % , 75 % de 2,3-diméthylbuta-1,3-diène (1) dans la 3,3-diméthyl butan-2-one (pinacolone) (2).
Préparer d'autre part 15 mL d'une solution B à 50 % de 4-méthylpentan- 2-one (I) dans (2).
Effectuer ensuite les mélanges suivants: aux 2 mL des solutions Ai, on ajoute 2 mL de B, opérations faites pour chacune des solutions Ai ainsi que pour (1) et (2) purs.
Injecter successivement les différents mélanges après avoir fait les règlages convenables.
température du four : 70°C
température de l'injecteur : 220°C
température du détecteur : 220°C
température du filament : 285°C
i = 200 mA
atténuation : 128
pression d'hélium : 6 bar
injection : 0,7 µL
Dans ces conditions, les temps de rétention observés sont :
(1) 2,3-diméthylbutadiène : 1,1 min
(2) 3,3-diméthylbutan-2-one : 2,2 min
(I) 4-méthylpentan-2-one : 2,9 min
Tracer la courbe d'étalonnage
Pour les solutions inconnues, ajouter à 1 mL de ces solutions 1 mL de B.
Déterminer le rapport des aires obtenues après injection.
A l'aide de la courbe d'étalonnage, déduire le pourcentage en (1) des solutions inconnues.
NB : La courbe d'étalonnage a été tracée à partir des solutions préparées ( % en volumes). Il est donc nécessaire d'en déduire les titres massiques correspondants.
titre massique en (1) =
UNE CHROMATOGRAPHIE QUE L'ON PEUT PORTER
Chromatographie sur T-shirt
Bibliographie
Journal of Chemical Education Décembre 92 p 877 et 978
L'exemple qui suit est une chromatographie radiale que l'on fera de préférence sous la hotte à cause des solvants utilisés:
Tendre sur un cristallisoir d'environ 15 cm de diamètre, un morceau de tissu de coton blanc. Le maintenir tendu à l'aide d'un élastique ou de ficelle.
Repérer le centre du cercle formé par le tissu.
Placer 4 à 6 taches de feutres permanents sur la circonférence d'un cercle imaginaire, environ quatre fois plus petit que le cristallisoir.
A l'aide d'un compte-gouttes, appliquer l'éluant (propan-2-ol/eau en proportions volumiques 7/3) au centre du cercle de telle sorte que l'éluant mouille le tissu et se déplace radialement vers les bords en créant un chromatogramme.
Quand le motif a atteint la taille voulue, arrêter les ajouts d'éluant et laisser sécher le tissu environ 10 min sous la hotte.