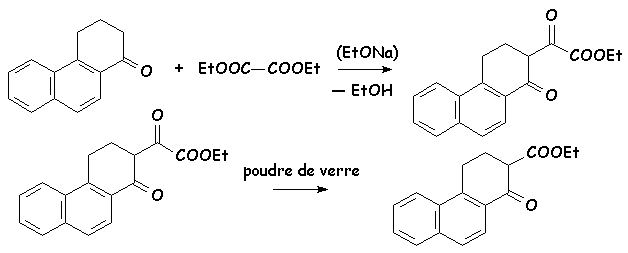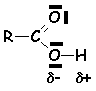
1. Structure. Propriétés physiques.
Les acides aliphatiques linéaires sont solides au delà de 10 C.
 Les acides carboxyliques se présentent sous une forme dimère à
létat pur ou en solution concentrée. En effet, la liaison hydrogène entre le
proton du groupement OH et loxygène du carbonyle dun autre groupement acide
carboxylique est très forte :
Les acides carboxyliques se présentent sous une forme dimère à
létat pur ou en solution concentrée. En effet, la liaison hydrogène entre le
proton du groupement OH et loxygène du carbonyle dun autre groupement acide
carboxylique est très forte :
Au point que la température de fusion de lacide correspond en fait à celle du dimère. Les acides aliphatiques sont liquides jusquà C10, puis solides (les cires de bougie par exemple). Les acides aromatiques sont tous solides.
Les diacides donnent souvent des liaisons H intramoléculaires :

Lacide maléique (ou but-2-ènedioïque Z) présente par exemple une température de fusion de 138°C, alors que lacide fumarique (ou but-2-ènedioïque E) fond à 287°C :
La liaison H est intramoléculaire dans le premier cas, et la Tf correspond à celle dun composé de masse molaire 116 qui crée 1 liaison H. Dans le second cas, on voit que le nombre de liaisons H intermoléculaires peut être très grand, doù la Tf très élevée.
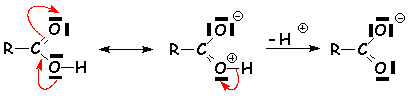
Cette liaison hydrogène très forte va affaiblir la liaison O H, autant que les effets mésomères attracteurs du carbonyle vont diminuer la densité électronique de loxygène. Ce proton est donc suffisamment mobile pour faire dun acide carboxylique un acide faible de Brønstedt. Les pK, pour les acides hydrocarbonés uniquement, varient de 3,7 pour lacide méthanoïque, à 4,9 pour lacide heptanoïque. Les acides substitués à proximité du groupement fonctionnel par des groupement attracteurs délectrons ont un PK plus faible (les effets attracteurs diminuent la stabilité de la liaison OH et augmentent donc lacidité) : pK =0 pour lacide trichloroéthanoïque. Par contre, les effets donneurs augmentent le pK. Lacide benzoïque a un pKa de 4,2
2. Réactivité des acides et de leurs dérivés.
Sur les schémas suivants, on voit les principaux sites réactionnels des acides et de leurs dérivés :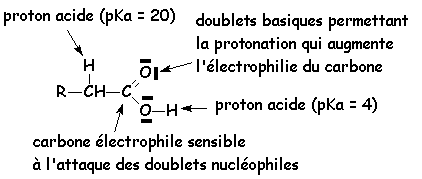
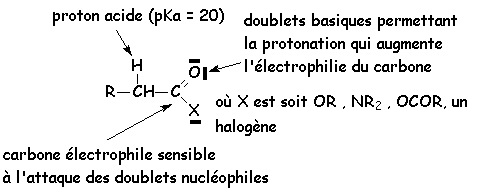
Si X º OR, cest un ester, X º NHmRn, cest une amide, X º halogène, cest un halogénure dacide, X º OCOR, cest un anhydride dacide.
Cas des nitriles :
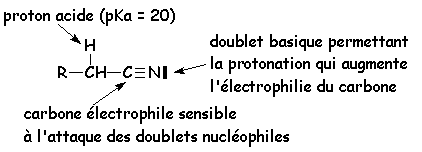
Tous les dérivés dacide sont hydrolysés en acide
Tous sont sensibles aux attaques nucléophiles
Les O des carbonyles et le N du nitrile peuvent être facilement protonés
Tous le H en a du carbonyle sont faiblement acide
3. Synthèse des dérivés dacide.
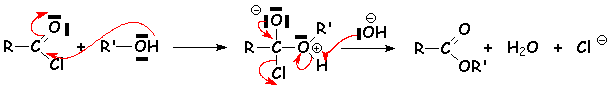
La réaction destérification est très connue. Cest une réaction équilibrée :
![]()
Le mécanisme est un mécanisme daddition élimination appelé SNAc2 :

On remarque bien que toutes les réactions sont équilibrées. La réaction est réversible. La réaction inverse est lhydrolyse de lester. Pour déplacer léquilibre dans le sens de formation de lester, on élimine leau grâce au dyn stark (voir la synthèse ses acétals) , dans un solvant tel que le benzène, en milieu acide tel que lacide paratoluènesulfonique, plus soluble que H2SO4 dans le benzène. Leau, léthanol, et le benzène forment un ternaire qui distille et libère leau lors de sa condensation.
Les acides réagissent complètement avec les agents halogénants classiques (PCl5, SOCl2) pour donner des chlorures dacide :![]()
![]()
Traités par P2O5, les acides sont déshydratés en anhydrides :
![]()
Les anhydrides sont aussi obtenus par réaction des ions carboxylates sur les chlorure dacide. Cela permet lobtention danhydrides mixtes :
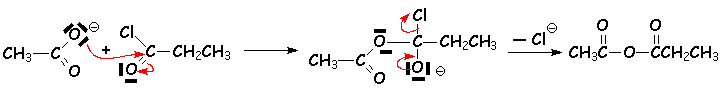
Les acides donnent des sels dammonium avec les amines :
![]()
Par chauffage, ce carboxylate dammonium se déshydrate :
![]()
4. Interconversion entre dérivés dacide.
|
Composés de départ | ||||||
|
Acide |
Ester |
Dérivé halogéné |
Anhydride |
Nitrile |
Amide | |
|
Acide |
+ H2O (+ H+) ou saponifica-tion suivie de protona-tion. |
Hydrolyse en milieu acide ou basique |
Hydrolyse en milieu acide ou basique |
Hydrolyse en milieu acide |
Hydrolyse en milieu acide ou basique | |
|
Ester |
+ alcool (+ H+) |
Transestérification |
+ ROH en milieu basique (NEt3) |
+ ROH |
+ ROH (1 mole + HCl sec) |
ROH (+ H+) |
|
Dérivé halogéné |
+ SOCl2 |
|||||
|
Anhydride |
+ P2O5 |
+ ion carboxylate (SNAC2) |
||||
|
Nitrile |
+ SOCl2 | |||||
|
Amide |
+ NH3 ou NH2R ou NHR2 (chauffage) |
+ NH3 ou NH2R ou NHR2 (H+) |
+ NH3 ou NH2R ou NHR2 (+ base NEt3) |
+ NH3 ou NH2R ou NHR2 |
H3O+ |
|
4.1. Hydrolyse en acide carboxylique.
La réaction dhydrolyse est la réaction inverse de la réaction destérification. Il suffit de chauffer lester avec un grand excès deau en milieu H2SO4 pour récupérer lacide et lalcool.
Mais cette réaction nest jamais totale, cest pourquoi, on préfèrera lhydrolyse en milieu basique
Lhydrolyse des amides se fait de manière semblable, mais elle est totale :
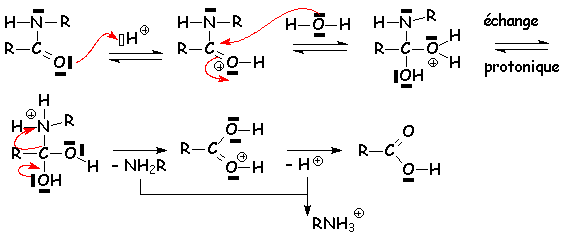
Les nitriles sont également hydrolysés totalement :
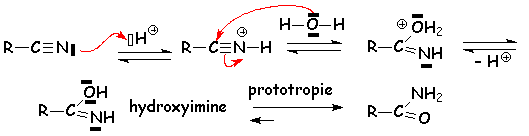
Lamide formée est hydrolysée comme indiqué
plus haut4.1.2. Catalyse basique Saponification.
Cette réaction chimique est une des plus anciennement utilisée, puisquelle permet la synthèse des savons. Lester est transformé à chaud de manière irréversible en carboxylate de sodium ou de potassium (le savon) et en alcool :

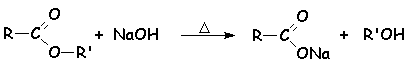
Le mécanisme est toujours une addition élimination :
Cest une réaction fort utilisée en synthèse, comme nous le verons plus loin, dautant que le passage en milieu acide permet de récupérer lacide carboxylique.
Les esters utilisés pour former les savons sont les esters dacides saturés et de glycérol, dont les principaux sont le tributanoate de glycérol (la butyrine, composant essentiel du beurre, le trihexadécanoate de glycérol (palmitine) et le trioctadécanoate de glycérol (stéarine), composants essentiels de lhuile darachide.
Lhuile dolive contient des esters dacides insaturés : oléine, linoléine, lhuile de lin la linolénine.
Voici les structures de tous les acides correspondants, les esters glycériques étant tous de la forme : RCOO CH2 CH(OCOR) CH2 OCOR
| Nombre de C | Structure | Nom usuel | Nomenclature | pKa |
| 1 | HCOOH | formique | méthanoïque | 3,77 |
| 2 | CH3COOH | acétique | éthanoïque | 4,76 |
| 3 | C2H5COOH | propionique | propanoïque | 4,88 |
| 4 | C3H7COOH | butyrique | butanoïque | 4,82 |
| 5 | C4H9COOH | valérique | pentanoïque | 4,81 |
| 6 | C5H11COOH | caproïque | hexanoïque | 4,85 |
| 7 | C6H13COOH | énanthique | heptanoïque | 4,85 |
| 8 | C7H15COOH | caprylique | octanoïque | 4,85 |
| 9 | C8H17COOH | pélargonique | nonanoïque | - |
| 10 | C9H19COOH | caprique | décanoïque | - |
| 12 | C11H23COOH | laurique | dodécanoïque | - |
| 14 | C13H27COOH | myristique | tétradécanoïque | - |
| 16 | C15H31COOH | palmitique | hexadécanoïque | - |
| 18 | C17H35COOH | stéarique | octadécanoïque | - |
| 22 | C21H43COOH | béhénique | doeicosanoïque | - |
| Acides insaturés : | ||||
| C8H17 CH=CH (CH2)7 COOH | oléïque | octadéc-9-énoïque | ||
| C5H11 CH=CH CH2 - CH=CH (CH2)7 COOH | linoléique | octadéca-9,12-diènoïque | ||
| C2H5 CH=CH CH2 CH=CH CH2 - CH=CH (CH2)7 COOH | linolénique | octadéca-9,12,15-triènoïque | ||
Les savons (carboxylates de sodium ou de potassium), présentent une extrémité hydrophile (la fonction carboxylate, par création de liaison H), et une extrémité lipophile (la chaîne carbonée linéaire).
Les matières grasses qui sont hydrophobes forment des sphères insolubles dans leau à cause des phénomènes de tension superficielle. Il ny a aucune liaison entre le corps gras et leau. Le savon, grâce à ses deux pôles, va créer un lien entre leau et le lipide, pour donner une émulsion permettant dentraîner le corps gras :
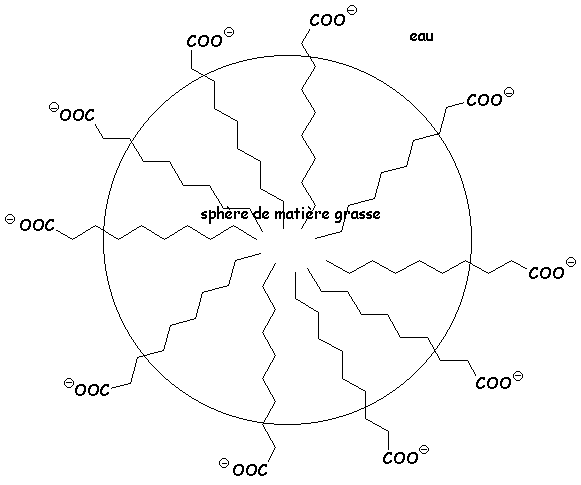
4.2. Obtention des esters à partir des dérivés dacide
4.2.1. Transestérification
Les esters peuvent être transformés en dautre esters, par chauffage avec un excès dalcool différent en milieu acide. Cette réaction permet par exemple de transformer les esters glycériques en esters éthyliques :
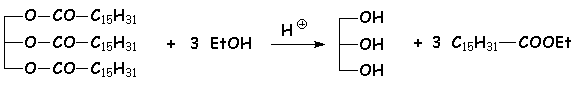
4.2.2. À partir des chlorures et anhydrides dacide.
Toute synthèse de dérivé dacide à partir de chlorure dacide nécessite la présence dune base car lion Cl formé au cours de la réaction est une base trop faible pour arracher les protons. Par contre lion carboxylate suffit pour cette opération, cest pourquoi les anhydrides se suffisent à eux-même :
Le mécanisme est toujours de même nature :
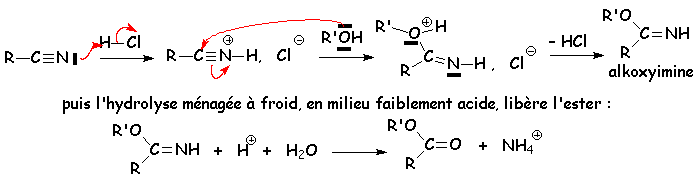
Cette réaction se fait en milieu acide et en présence dun excès dalcool. Elle est semblable à la réaction destérification à partir des acides :
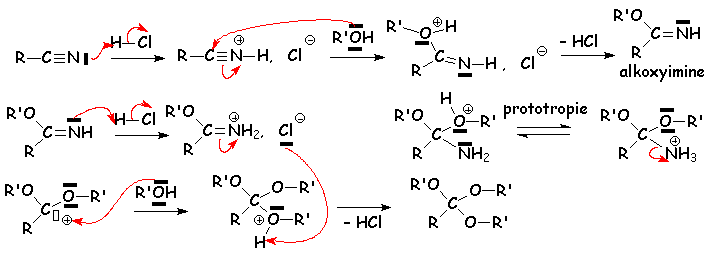
Une mole dalcool réagit avec une mole de nitrile en présence de gaz chlorhydrique sec pour donner un ester. Par contre, trois moles dalcool donneront, toujours avec une mole de nitrile dans les mêmes conditions, un orthoester RC(OR)3 :
Mais en présence dun excès dalcool :
Cest la réaction inverse de la synthèse des esters vue précédemment. Les conditions opératoires sont les mêmes. On se place en excès damine ou dammoniac.
4.2.6. À partir des chlorures dacide.
Généralement, au laboratoire, on forme les amides de cette manière. La réaction doit se faire en milieu basique (soit de la soude diluée, soit une amine tertiaire comme la pyridine ou la triéthylamine) :
Comme toujours en catalyse basique, cest le nucléophile (ici lamine) qui attaque le carbonyle :
4.2.7. À partir des anhydrides
Comme dhabitude, les anhydrides réagissent avec les nucléophiles sans quil y ait besoin de base :
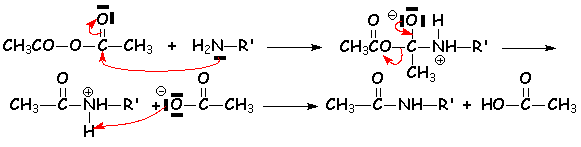
5. Réduction des acides et de leurs dérivés.
5.1. Acides.
Les acides sont très stables, aussi sont-ils difficiles à réduire. Les hydrures réagissent généralement pour donner un dégagement dhydrogène :
![]()
Le tétrahydruroaluminate de lithium LiAlH4 donne le même composé intermédiaire, mais, lorsque celui-ci ne précipite pas, LiAlH4 va réduire le carboxylate en alcoolate, il y a donc formation dalcool après hydrolyse :
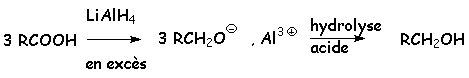
Cette réaction est donc aléatoire, et on préférera, pour réduire les acides carboxyliques en alcools, transformer ceux-là en esters, facilement réduits en alcools par LiAlH4 (§ 5.4.1.)
Ils sont réduits en alcools par LiAlH4, puis hydrolyse acide. Mais leur intérêt réside dans leur transformation en aldéhyde :
Le traitement dun chlorure dacide par lhydrogène en présence de palladium désactivé par du soufre et du sulfate de baryum permet dobtenir un aldéhyde :
5.2.2. Utilisation du tritertiobutoxyhydruroaluminate de lithium
Ce réducteur donne le même résultat après hydrolyse acide de lintermédiaire obtenu :
Les dérivés azotés des acides (amides et nitriles) donnent toujours des amines par réduction avec LiAlH4 ou H2/Ni (ou Pt). La groupement carbonyle est transformé en méthylène CH2 . C º N est réduit en CH2 NH2
Les esters sont très facilement réduits en deux alcoolates par LiAlH4. Lhydrolyse conduit à deux alcools :
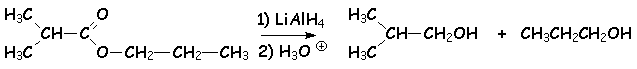
5.4.2. Réaction de Bouvault et Blanc.
Les esters sont réduits par le sodium de manière radicalaire. En présence de protons, tels que ceux amenés par le solvant non aqueux qui peut être léthanol, ils sont réduits en deux alcools, comme dans la réaction précédente :
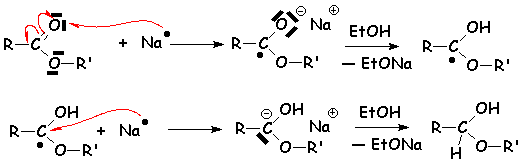
Lhémiacétal formé est instable et se décompose en aldéhyde R CH = O et en alcool ROH. Laldéhyde est ensuite réduit de la même manière.
Voici léquation bilan de la réduction dun ester en milieu protique (éthanol) par le sodium métallique :
![]()
Lhydrolyse acide qui suit détruit les alcoolates et libère les alcools.
Si lester réagit avec le sodium en labsence de solvant protique (dans le toluène par exemple), une duplication a lieu (voir la duplication réductive des cétones par les métaux). On obtient des acyloïnes, composé carbonylé a-hydroxylé :
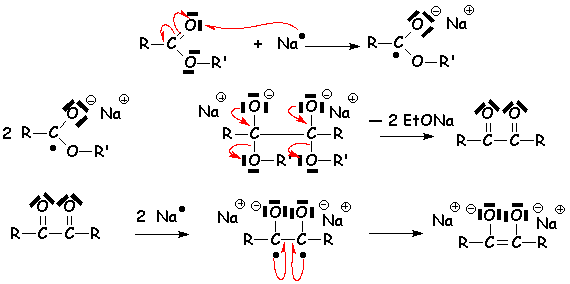
Lors de lhydrolyse acide qui suit, les oxoanions sont protonés et le composé se tautomérise en acyloïne :
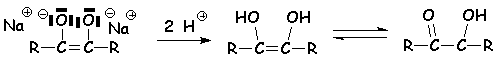
5.4.4. Réduction industrielle.
5.5. Action des organométalliques
Le tableau ci-dessous résume les réactions des acides et dérivés avec les organométalliques principaux.
À remarquer les 5 méthodes permettant de synthétiser des cétones en utilisant des organométalliques.
Les orthoesters subissent une seule SN2 où EtO est substitué par R, et lhydrolyse acide du cétal formé (stable en milieu basique) donne la cétone
|
Organométallique Dérivé dacide |
Organomagnésien RMgX |
Organolithien RLi |
Organocadmien R2Cd |
|
Acide RCOOH |
RCOOMgX+ + RH
|
|
/ |
|
Ester RCOOR |
|
|
|
|
Halogénures RCOCl |
Id |
Id |
R CO R |
|
Anhydrides RCOOCOR |
Id |
Id |
|
|
Amide I RCONH2 |
RCONHMgX+
|
||
|
Amide III RCONR2 |
|
||
|
Nitrile RCN |
|
||
|
Orthoester RC(Oet)3 |
|
6. Réactivité des hydrogènes en
a du carbonyle. 6.1. Halogénation des acides en a : réaction de Hell-Volhard-Zelinski.Il sagit de lhalogénation sur le carbone en a de la fonction acide.
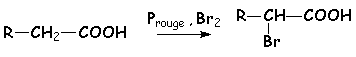
Le Phosphore rouge est en quantité catalytique, il réagit avec le brome pour donner PBr3 qui va permettre de créer une faible quantité de bromure dacyle RCH2COBr. Les effets I du brome vont augmenter lacidité des H en a du carbonyle, et permettre la formation de lénol. Comme avec les dérivés carbonylés, il y a alors addition de dibrome, avec élimination dHBr. Puis les fonctions acide et bromure dacide séchange, en régénérant une quantité catalytique de bromure dacyle :
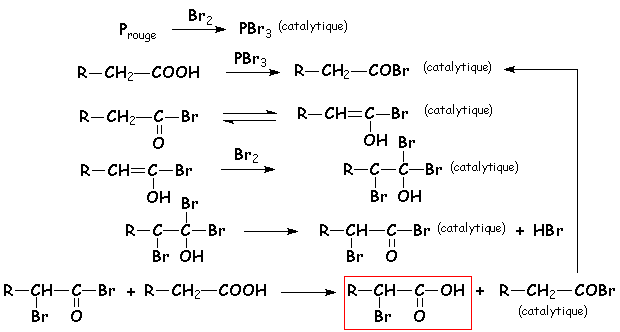
Cette réaction permet de rajouter deux carbones à une chaîne carbonée halogénée, dont un représente une fonction acide carboxylique. La suite des réactions se décrit ainsi :
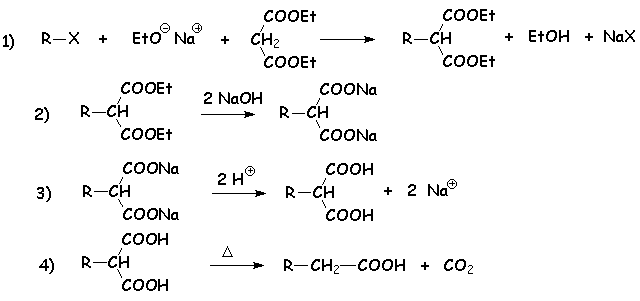
La première réaction débute par larrachement du proton porté par le CH2 du propanedioate déthyle (appelé malonate déthyle). Le carbanion formé est stabilisé par laddition des effets mésomères et inductifs attracteurs des deux fonctions esters, doù cette acidité relativement élevée.
Ce carbanion opère ensuite une SN2 sur le dérivé halogéné.
On transforme ensuite le diester en diacide par une saponification suivie dune acidification. Le chauffage de ce diacide conduit à une décarboxylation, classique pour des acides b,g-insaturés.
6.3.1. En b de C=O
Cest une réaction à 6 centres :
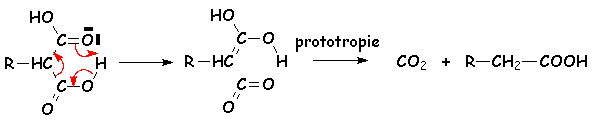
Les b-halogénocarboxylates se décarboxylent aussi par chauffage :
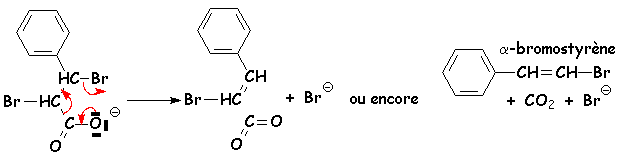
Les deux groupes partant (CO2 et Br) doivent être en anti pour que l'élimination se fasse bien (réaction stéréospécifique)
Tous les acides peuvent être décarboxylés par décomposition radicalaire des peresters, obtenus par estérification de lacide par un hydroperoxyde dalkyle, dans un solvant donneur de H·, tel que le cumène :
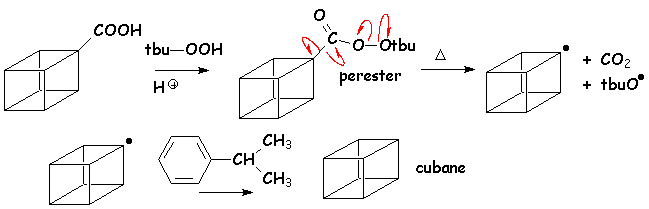
6.4. Condensation de Claisen et réactions apparentées.
En présence déthanolate (ou éthylate) de sodium en quantité catalytique à chaud, les esters possédant un hydrogène en a se condensent :
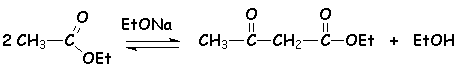
Le composé obtenu ici est le 3-oxo-butanoate déthyle (appelé lacétylacétate déthyle). Le mécanisme est toujours une addition élimination :
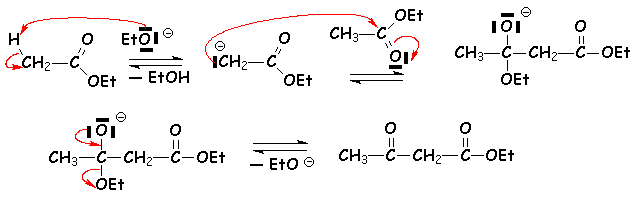
Cette réaction est équilibrée, et la formation de lacétylacétate déthyle est obtenue par distillation de léthanol. Si lon chauffe lacétylacétate déthyle avec un excès déthanol en présence dune quantité catalytique déthanolate de sodium, on récupère léthanoate déthyle de départ. Par contre la saponification de lacétylacétate déthyle suivie de chauffage en milieu acide conduit à une décarboxylation (acide b,g-insaturé). Il se forme la propanone.
6.4.2. Synthèse acétylacétique
On peut utiliser ces propriétés pour mettre en uvre une suite de réaction semblable à la synthèse malonique : cest la synthèse acétylacétique.
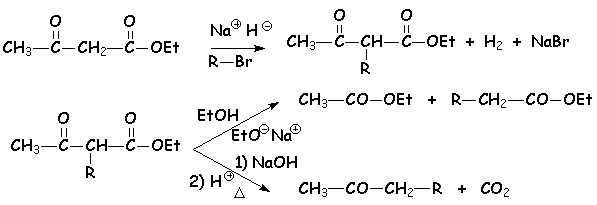
Comme indiqué plus haut, il y a deux façon de traiter lester obtenu. Cette réaction permet donc dobtenir, soit un ester substitué, soit une cétone substituée.
Les 3 réactions de cyclisation qui vont suivre, sont des réactions de type Claisen, suivies de décarboxylation.
On utilise un diester, traité par une base forte : NaOEt, NaH, [(CH3)2CH]2NNa (diisopropylamidure de sodium), Na lui-même ( il y a dégagement dhydrogène) :
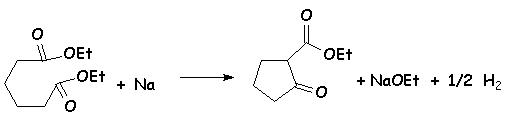
Le mécanisme est le même que celui de la condensation de Claisen
La saponification, suivie dun chauffage du sel formé en milieu acide, conduit à la cyclopropanone par décarboxylation :
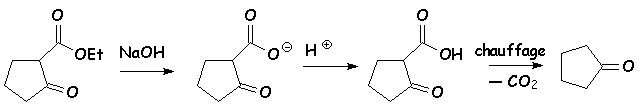
Le chauffage dun diacide carboxylique avec de lhydroxyde de baryum (baryte) conduit au même résultat que précédemment. Le moteur de la réaction semble être la formation de carbonate d baryum très stable (cf son insolubilité dans leau) :
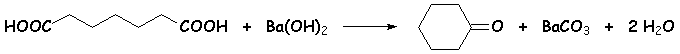
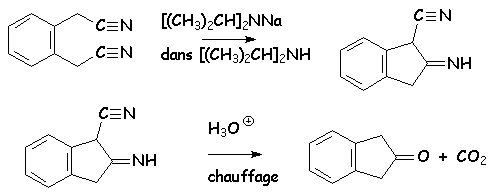
Elle est tout à fait semblable à la Dieckmann. Au lieu du diester, on utilise le dinitrile. Celui-ci est traité par le diisopropylamidure de sodium par exemple, pour donner, dans la diisopropylamine comme solvant, une a-cyanoimine. Lhydrolyse transforme le groupement nitrile en acide carboxylique, limine en cétone, et le chauffage simultané permet alors la décarboxylation avec libération de la cétone cyclique :
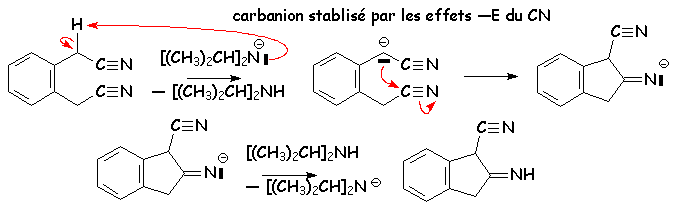
Le mécanisme en est classique :
6.5. Applications. Réaction des esters et autres dérivé dasside avec les aldéhydes et cétones.
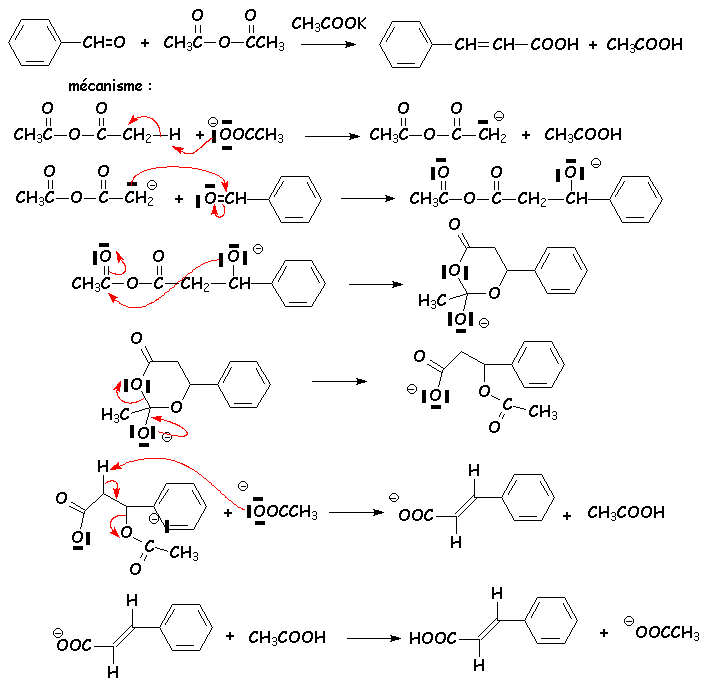
Les trois premières réactions montrent une diminution de lacidité des H en a du CO du dérivé dacide, vérifiée par laugmentation de la force de la base utilisée (ion éthanoate dans le cas de la Perkin, soude pour la Knævenagel, éthanolate pour la Stobbe). Les deux pemières sont dailleurs des équilibres favorisés vers la formation de composés fortement conjugués et donc très stables.
Le benzaldéhyde réagit avec lanhydride éthanoïque en présence dion éthanoate. Les effets attracteurs des deux carbonyles se cumulent pour rendre acide lhydrogène en a de lun ou lautre des C=O :
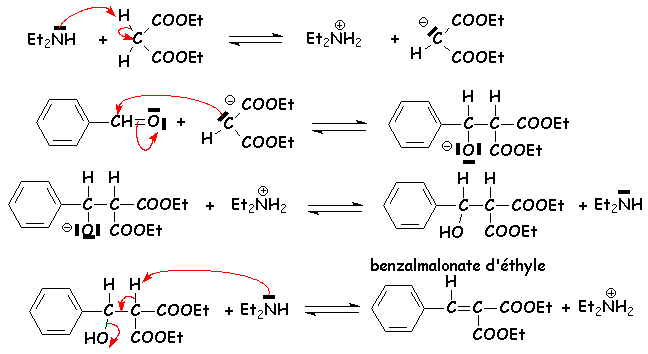
Le malonate déthyle est suffisamment acide pour quune base assez forte telle que la soude ou une amine secondaire est capable de permettre la mise en place de la réaction équilibrée avec le benzaldéhyde qui sera déplacée vers la droite par élimination deau et formation dun composé encore très conjugué :
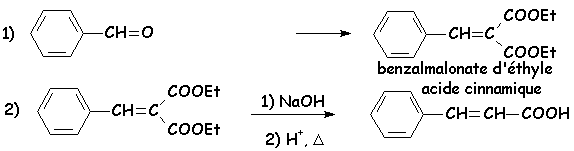
Le mécanisme est semblable à la synthèse malonique. Voilà pour la première partie :
Le succinate déthyle (butanedioate déthyle) est beaucoup moins acide que les deux composés précédents (anhydride ou malonate). Cependant, les effets inductifs des deux fonctions ester sont suffisants pour que un des protons en a dune des fonctions esters soit plus acide quun proton en a dun dérivé carbonylé. Celui dailleurs possèdent, comme dans les deux cas précédents, le carbone le plus électrophile :
6.5.4. Utilisation de loxalate déthyle
Contrairement aux réactifs précédents, loxalate déthyle (éthanedioate déthyle) ne possède pas dhydrogène acide. Par contre, la proximité des deux fonctions esters, et de leurs effets attracteurs (inductifs essentiellement), rend le carbone fonctionnel plus électrophile que celui dun dérivé carbonylé classique. Sur lexemple suivant, on obtient en milieu éthylate un ester a-carbonyle, que lon décarbonyle ( CO) ensuite par chauffage sur poudre de verre. Cette réaction sert à fixer un carboxylate déthyle en a dun carbonyle