Cet effet intéresse les doublets . Lorsque la liaison est formée entre deux éléments de nature différente, les électrons sont attirés vers l'élément le plus électronégatif.
On appelle ”effet inducteur négatif” -I les effets résultant du déplacement des électrons d'une liaison vers un atome plus négatif que le carbone, et effet inducteur positif +I les effets résultant de leur déplacement vers le carbone en s'éloignant d'un atome moins électronégatif que lui :
![]()
Ces effets sont à l'origine de la polarisation de la molécule. Un moment dipolaire apparaît : exemple des halogènes
| F | Cl | Br | I | |
| 1,81 | 1,87 | 1,70 | 1,59 | |
| 1,92 | 2,05 | 2,02 | 1,90 | |
| 2,10 | 2,15 | 2,01 | ||
| 2,09 | 2,15 | 2,08 | ||
| 2,12 |
1.1. Effet inducteur négatif -I.
Ceux-ci sont très forts pour les molécules présentant des charges positives complètes ; ils sont d'autant plus intenses que l'atome lié à C est plus électronégatif :
Dans ![]() , l'effet est plus intense que
dans
, l'effet est plus intense que
dans ![]() , lui-même plus fort que dans
, lui-même plus fort que dans ![]() .
.
Les effets restent forts même quand la charge est compensée par une charge négative voisine (liaison semipolaire) :
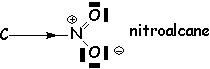
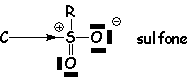
Pour les atomes non chargés, l'effet dépend de leur électronégativité :
![]()
Les effets inductifs sont additifs :
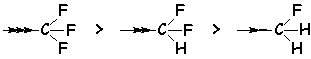
On trouve de même un effet inductif extrêmement fort pour :
![]()
Les groupes fonctionnels qui donnent lieu à une mésomèrie et dont au moins une forme limite présente une charge + exercent également un effet -I assez important :
![]()
L'état d'hybridation a aussi une influence :
![]()
1.2. Effet inductif positif +I.
Les atomes chargés + ont un fort effet +I : celui-ci decroît quand l'électronégativité croît.
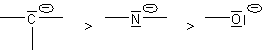
Plus l'atome non chargé est électropositif, plus l'effet +I est important :
![]()
Les groupes alkyles ont un effet +I. Plus ils sont ramifiés, plus cet effet est important :
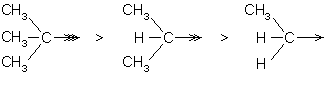
2.1. Structure.
Elle est pyramidale (![]() ) et l'inversion de configuration y
est très rapide :
) et l'inversion de configuration y
est très rapide :

Par contre les radicaux vinyliques ne se racémisent pas :
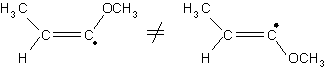
| Ed | Ed | ||
| 426 | 376 | ||
| 426 |  |
326 | |
| 410 | 324 |  | |
| 393 | 322 |
Ils sont stabilisés par les effets donneurs, par résonance :
![]()
On peut évaluer la stabilité des
radicaux ![]() par l'énergie de dissociation (en
par l'énergie de dissociation (en ![]() ) de C-H :
) de C-H :
Les radicaux les plus stables sont ceux pour lesquels Ed est faible.
2.3. Modes de formation essentiels
2.3.1. Par
addition d'un ![]() à une liaison C==C :
à une liaison C==C :
![]()
2.3.2. Par rupture homolytique de certaines structures particulières :
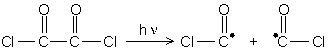

azobisisobutyronitrile :

2.3.3. Par réaction d'une liaison C-H avec un radical
hétéroatomique (le plus souvent ![]() ) :
) :
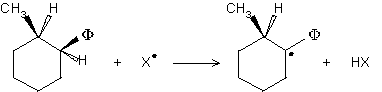
2.4. Évolution des radicaux libres.
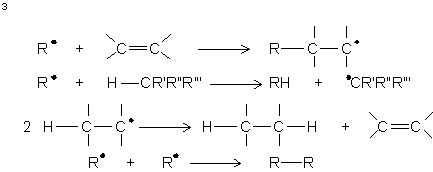
Ce sont des composés avides d'électrons
(électrophiles). Ils réagiront avec des structures non déficientes en électrons
: doublet , doublets ou encore couplage (avec ou sans dismutation) de deux
radicaux :
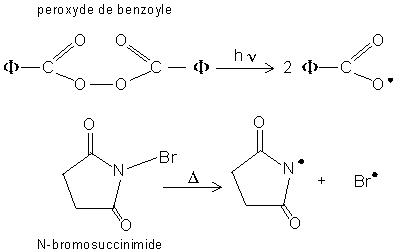
2.5. Radicaux libres hétéroatomiques.
Ils sont formés par rupture homolytique :
![]()
Ils ne sont pas isolables (de même que les radicaux carbonés). Ils ont un caractère électrophile et attaquent aussi les doublets et . Pour les former, il suffit de chauffer ou d'irradier les composés présentant des liaisons hétéroatome-hétéroatome.
3. Réactions d' oxydation des alcanes.
![]() .Cette réaction libère de l'énergie (combustion des dérivés du
pétrole). Elle permet également la connaissance des énergies de liaison pour
C--C et C--H . C'est une réaction radicalaire :
.Cette réaction libère de l'énergie (combustion des dérivés du
pétrole). Elle permet également la connaissance des énergies de liaison pour
C--C et C--H . C'est une réaction radicalaire :

initiation :
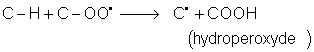
propagation :
![]()
ramification :
C'est une réaction en chaînes
ramifiées, ce qui entraîne une explosion. Il existe quelques réactions
industrielles d'oxydation contrôlée : celle du cumène ![]() en propanone
en propanone ![]() et en phénol
et en phénol ![]() .
.
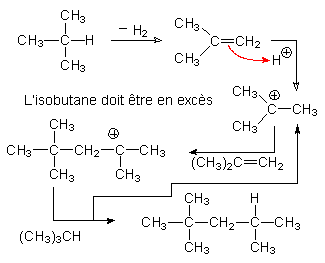
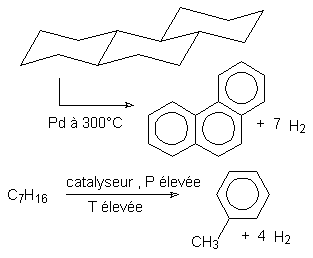
Plus un alcane est ramifié, plus élevée sera sa pression d'explosion en présence d'oxygène. De la mesure de cette pression, on tire l'indice d'octane d'un hydrocarbure : i.o. = 0 pour le n-octane, i.o. = 100 pour le 2,2,4-triméthylpentane.
Il est donc intéressant d'isomériser les alcanes en chaînes ramifiées. L'initiateur est un carbocation obtenu par action de H ; sur un alcène ou sur un chlorure d'alkyle en présence d' AlCl3 .
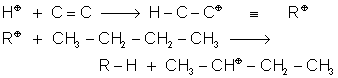
Il y a ensuite Réarrangement de WAGNER-MEERWEIN
![]()
Puis le carbocation tertiaire obtenu réagit avec l'alcane de départ :
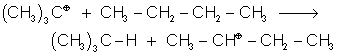
exemple :
![]()
![]()
Le catalyseur utilisé : Chrome +
Al2O3 . C'est l'alumine qui permet
la formation de ![]() .
.
C'est un procédé industriel :
Intéressant pour obtenir industriellement les aromatiques :
8.1. Notion de mécanisme réactionnel.
8.1.1. États de transition.
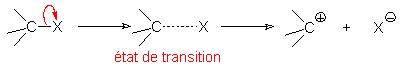
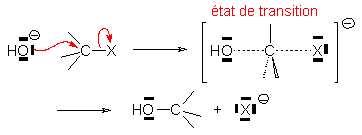
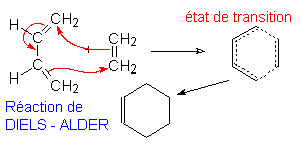
Les états de transition représentent les points où l'évolution d'une réaction est réversible. On représente alors les liaisons qui se forment ou se rompent par des traits pointillés, qu'il s' agisse d'attaque ou de départ, que plusieurs liaisons se forment ou se rompent simultanément ou non.
8.1.2. Étude thermo-dynamique de la réaction.
Considérons la réaction équilibrée
![]()
On sait que G de la réaction est égal
à : G = - RT. ln K avec  . Pour une réaction instantanée et spontanée, les variations de
G en fonction du
degré d'avancement de la réaction sont représentées ainsi :
. Pour une réaction instantanée et spontanée, les variations de
G en fonction du
degré d'avancement de la réaction sont représentées ainsi :
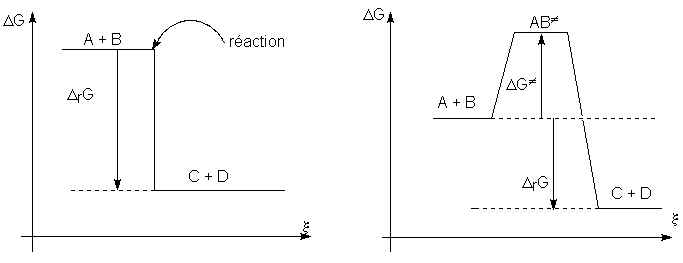
Cependant le premier cas n'existe pratiquement jamais. Les réactions s'effectuent généralement avec passage par un maximum d'énergie, comme sur la seconde figure.
Pour que la réaction se produise, on doit
fournir aux réactifs une énergie supplémentaire pour ”activer” les molécules :
c'est l'énergie d'activation ![]() . Le maximum de la courbe correspond à l'état de transition de la
réaction ou complexe activé. Ce n'est pas un intermédiaire isolable. À partir de
cet état de transition, le système peut aussi bien progresser que régresser, son
énergie diminuant dans les deux cas.
. Le maximum de la courbe correspond à l'état de transition de la
réaction ou complexe activé. Ce n'est pas un intermédiaire isolable. À partir de
cet état de transition, le système peut aussi bien progresser que régresser, son
énergie diminuant dans les deux cas.
On peut alors représenter la réaction par le schéma suivant :
![]()
8.1.3. Vitesse de la réaction chimique.
La vitesse d'une réaction chimique élémentaire est égale , par définition, à la dérivée de la concentration d'une espèce (de l'état initial ou de l'état final) par rapport au temps, affectée de l'inverse du coefficient stoechiométrique et du signe ”moins” s'il s'agit d'un réactif :
![]()
L'expérience montre que cette vitesse
v est aussi égale à ![]() ,
k1 étant la
”constante de vitesse” de la réaction, k1 étant relié à
,
k1 étant la
”constante de vitesse” de la réaction, k1 étant relié à ![]() par la relation
suivante :
par la relation
suivante : ![]() .
.
8.1.4. Intermédiaires réactionnels.
Une transformation peut se faire en
plusieurs étapes simples, avec apparition de plusieurs intermédiaires, par
exemple la substitution nucléophile SN1 d'un halogène par ![]() :
:
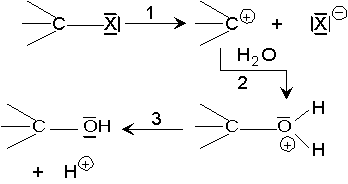
B et C sont des intermédiaires réactionnels.
Le diagramme énergétique de cette réaction
est le suivant :
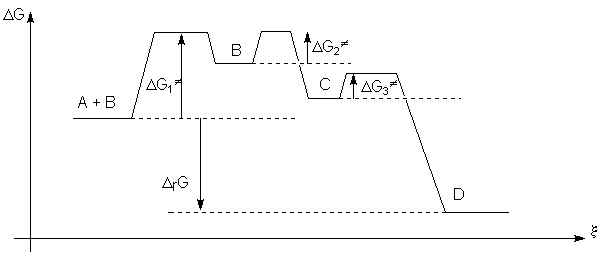
Ici ![]() est pratiquement nul. La
réaction 3 a une vitesse quasi infinie.
est pratiquement nul. La
réaction 3 a une vitesse quasi infinie. ![]() est très petit. La vitesse de
cette réaction est très grande. Dès que le premier intermédiaire réactionnel, B,
est formé, la réaction 2 a lieu. Par contre
est très petit. La vitesse de
cette réaction est très grande. Dès que le premier intermédiaire réactionnel, B,
est formé, la réaction 2 a lieu. Par contre ![]() est très élevé. La vitesse de
cette réaction est faible. C'est elle qui globalement imposera sa vitesse à la
totalité de la réaction. Comme la première étape est
monomoléculaire,
est très élevé. La vitesse de
cette réaction est faible. C'est elle qui globalement imposera sa vitesse à la
totalité de la réaction. Comme la première étape est
monomoléculaire,
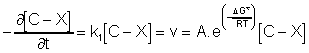
il arrive qu'une autre réaction ait
un ![]() plus important, c'est elle qui limite
alors la réaction.
plus important, c'est elle qui limite
alors la réaction.
8.1.5. Contrôle cinétique et thermodynamique d'une réaction.
Soit deux réactions ![]() et
et ![]()
Si les conditions expérimentales et le temps de la réaction sont tels que ces deux équilibres ne peuvent s'établir, c'est le produit obtenu par la réaction la plus rapide qui sera isolé en majorité :
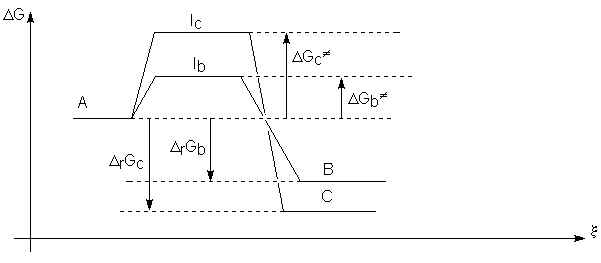
Ici, cela sera B, car ![]() est le plus petit.
Par contre, si on laisse les équilibres s'établir (par exemple en augmentant la
température) le produit majoritaire sera C , car
est le plus petit.
Par contre, si on laisse les équilibres s'établir (par exemple en augmentant la
température) le produit majoritaire sera C , car ![]() est le plus
négatif (loi d'action de masse). Le premier cas correspond à un contrôle
cinétique de la réaction, le second a un contrôle thermodynamique.
est le plus
négatif (loi d'action de masse). Le premier cas correspond à un contrôle
cinétique de la réaction, le second a un contrôle thermodynamique.
Si le contrôle est cinétique, la
proportion des produits B et C dépend de la différence ![]() :
:
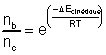 qui est donc supérieur à
1.
qui est donc supérieur à
1.
Si le contrôle est thermodynamique,
cette proportion dépend de ![]() :
:
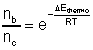 qui est donc inférieur à 1.
qui est donc inférieur à 1.
Exemple :
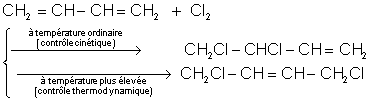
8.2. Mécanisme de la réaction d' halogènation.
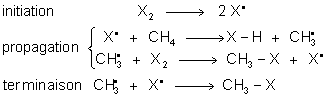
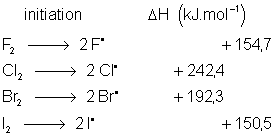
On constate une formation plus difficile
des radicaux ![]() .
.
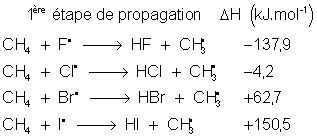
La propagation est pratiquement stoppée pour la bromation et l'iodation, sauf lorsque le substrat est tellement réactif que l'enthalpie de la réaction devient négative :
![]()
![]()
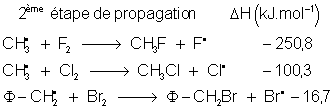
On peut donc conclure que
F2 et
Cl2 réagiront quel que soit
le substrat, avec une préférence tout de même pour ceux donnant des radicaux
stables (surtout pour ![]() ) : ils sont peu sélectifs. Par contre
) : ils sont peu sélectifs. Par contre ![]() ne réagira qu'avec des substrats
donnant des radicaux très stables : il est très sélectif. Étudions maintenant
les produits obtenus.
ne réagira qu'avec des substrats
donnant des radicaux très stables : il est très sélectif. Étudions maintenant
les produits obtenus.
8.2.2. Produits d'halogènation.
Le pourcentage des différents isomères obtenus dépendra de la sélectivité de l'halogène et aussi de la proportion de chaque type de H dans la molécule. Par exemple le 1-méthyl-propane comporte 9 H primaires et un H tertiare ; statistiquement, on devrait obtenir neuf fois plus de 1-chloro-2-méthyl-propane (I) que de 2-chloro-2-méthyl-propane (II).
Expérimentalement, on obtient deux fois plus de (I) que de (II). La position tertiaire est donc plus réactive que la primaire, et on constate qu'elle l'est 4,5 fois plus :
| réactivité
relative |
proportion
|
Avec Br2, très sélectif, on n'obtient pratiquement que du 2-bromo-2-méthyl-propane.
Les transpositions ou réarrangements (passage d'un groupe d'un carbone à un autre) ne se font pas avec les radicaux :
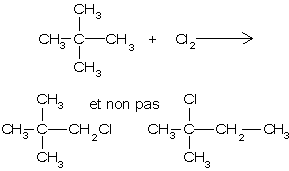
En présence d'un grand excès d'alcane, on obtient le dérivé monohalogèné. Par contre, avec les proportions stœchiométriques, on obtient un mélange :

Ces réactions sont habituellement
initiées par le rayonnement électromagnétique ![]() , mais aussi par des peroxydes
, mais aussi par des peroxydes
![]() ,
des composés azoïques tels que l'azobisisobutyronitrile, le chlorure de
sulfuryle SO2Cl2 .
,
des composés azoïques tels que l'azobisisobutyronitrile, le chlorure de
sulfuryle SO2Cl2 .
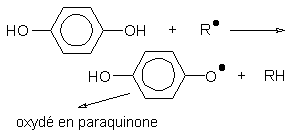
Les inhibiteurs bloquent les réactions radicalaires, par exemple l'hydroquinone :
9. Insertion de carbènes dans l'alcane
9.1. Étude des carbènes CZ2
9.1.1. Structure.
Les carbènes présentent un carbone hybridé sp2 comportant deux paires de liaison, une paire sp2 libre, et une lacune électronique portée par l'OA pz perpendiculaire aux deux liaisons :
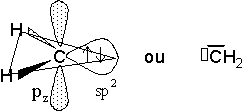
9.1.2. Obtention des carbènes.
Obtenus principalement par élimination géminée des dérivés halogénés des hydrocarbures. Les plus stables sont ceux soumis à des effets donneurs :
![]()
9.1.2.1. Déshydrohalogènation.
Les dérivés halogènés présentant un H
mobile peuvent donner des carbènes sous l'action de bases fortes, telles que les
carbanions ![]() ou les amidures
ou les amidures ![]() .
. ![]() :
:
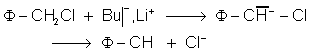
Les dihalogènocarbènes s'obtiennent par action d'une base alcaline (NaOH, KOH) sur un haloforme (CHX3).
![]()
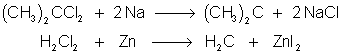
9.1.3. Réactivité des carbènes.
Ils sont fortement électrophiles par leur lacune et attaquent les doublets et :
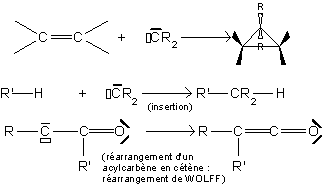
9.2. Réaction des carbènes avec les alcanes.
Il y a insertion du carbène entre C et H . Cette réaction ne présente aucune sélectivité et les proportions d'isomères obtenus dépendent du nombre de chaque type d'hydrogène (effet statistique) :
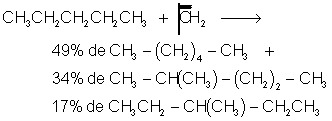
![]()
10.2. Hydrogènation des alcènes et des alcynes.
![]()
10.3. Réductions de WOLFF-KISCHNER et de CLEMMENSEN.
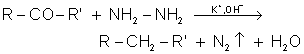
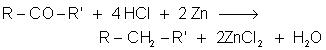
10.4. Réduction des dérivés halogènés par LiAlH4 ou par H2.
![]()
10.5. Réduction des halogènures par Mg puis hydrolyse.
