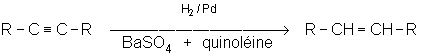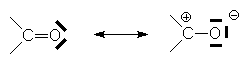
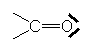
Si l’on considčre les deux formes limites d’un carbonyle :
la contribution de la forme (II) détermine
un déplacement partiel du doublet p
vers O . Cet effet mésomčre n’est important que dans une structure oů le groupe
accepteur (C=O) est conjugué avec un groupe donneur d’électrons  :
: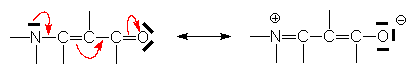
Les effets mésomčres sont négatifs lorsqu’il y a acceptation d’électrons (C=O), et positifs lorsqu’il y a don d’électrons
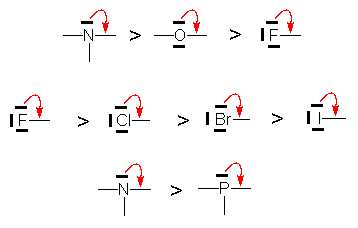
Les atomes présentant un doublet libre peuvent le donner pour former une liaison p avec l’atome voisin. Cette tendance diminue dans une męme période avec l’augmentation de l’électronégativité, mais, dans une męme colonne, diminue avec l’augmentation de la taille de l’atome.
Les groupes alkyles présentent un effet +E par hyperconjugaison. Cet effet diminue lorsque la substitution augmente sur le carbone.
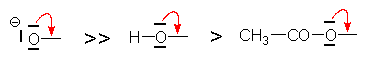
Les charges négatives confčrent un trčs fort effet +E. La substitution d’un groupe +E par un groupe -E diminue l’effet +E du premier
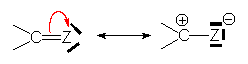
Les fonctions insaturées peuvent accepter un doublet, par conversion de leur double liaison en un doublet n :
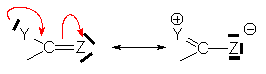
Cette tendance est d’autant plus forte que l’atome accepteur est plus électronégatif :
 >
> ![]()
Cet effet est considérablement augmenté si l’atome
possčde une charge ![]() :
:
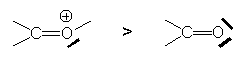
Il est diminué s’il est compensé par conjugaison avec un groupe donneur
Ceci rend compte de la diminution de l’effet –E dans la série :
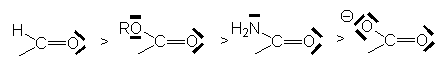
Autre exemple important : 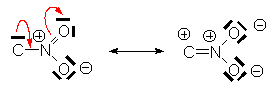
Les effets E se transmettent par les systčmes p conjugués, alors que les effets I se transmettent le long des liaisons s .
groupement |
– E |
– I |
+ I |
+ E |
|
´ ´ ´ ´ |
´ ´ ´ ´ |
||
|
´ ´ ´ |
´ ´ ´ |
||
|
´ ´ ´ ´ |
|||
|
´ ´ ´ ´ |
|||
|
´ ´ |
´ ´ |
||
|
´ ´ |
´ ´ ´ |
||
|
´ |
´ ´ |
||
|
´ ´ ´ |
|||
|
´ ´ ´ |
´ ´ |
||
|
´ ´ |
´ |
||
|
´ |
´ |
||
|
´ |
´ |
´ |
|
|
´ |
´ |
||
|
´ ´ |
|||
|
´ |
|||
|
´ ´ |
|||
|
´ ´ ´ |
|||
|
´ ´ ´ |
|||
|
´ ´ ´ |
´ ´ ´ ´ |
||
2. Structure de la double liaison.
L’énergie de liaison vaut 606 ![]() . Grâce ŕ la méthode OM (voir infra), on peut
déterminer l’énergie de la liaison p (b
pour une molécule,
Nb pour une mole). On trouve 188
. Grâce ŕ la méthode OM (voir infra), on peut
déterminer l’énergie de la liaison p (b
pour une molécule,
Nb pour une mole). On trouve 188 ![]() pour
pour ![]() . La liaison
s faisant partie de cette double liaison a donc une énergie de 418
. La liaison
s faisant partie de cette double liaison a donc une énergie de 418
![]() .
.
On remarque que cette énergie est plus élevée que celle
d’une liaison s classique (347 ![]() ). Ceci est lié au
raccourcissement de la liaison : 0,1544 nm pour C—C et 0,1334 nm pour C=C. La
liaison s est donc trčs solide,
alors que la liaison p est facile ŕ
rompre : c’est elle qui fait la réactivité des alcčnes.
). Ceci est lié au
raccourcissement de la liaison : 0,1544 nm pour C—C et 0,1334 nm pour C=C. La
liaison s est donc trčs solide,
alors que la liaison p est facile ŕ
rompre : c’est elle qui fait la réactivité des alcčnes.
Ce doublet p de liaison est riche en électrons, qui sont relativement peu retenus par les atomes de carbone. Ces électrons seront donc trčs facilement sollicités par les groupements pauvres en électron, dits électrophiles. Le doublet p a donc des propriétés nucléophiles importantes. Comme on étudie généralement l’action des agents extérieurs sur l’alcčne, on parlera d’additions et de substitutions électrophiles sur la double liaison.
3. Nomenclature et propriétés physiques.
On remplace le suffixe -ane par le suffixe -čne.
Quelques noms usuels : propylčne pour propčne ![]() . Les cycloalcčnes trans
n’existent que si
. Les cycloalcčnes trans
n’existent que si ![]() .
La nomenclature est un peu particuličre pour certains cycles :
.
La nomenclature est un peu particuličre pour certains cycles :
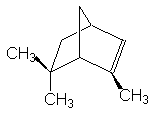
a -pinčne ou 2,6,6-triméthyl-bicyclo [2,2,1] hept-2-čne
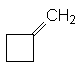
méthylčnecyclobutane
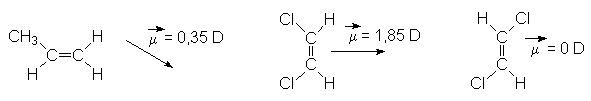
3.2. Moment dipolaire des alcčnes.
Ŕ cause de la rigidité de la molécule, il existe un moment dipolaire pour les molécules dissymétriques :
Ainsi qu’on le voit en spectroscopie, la circulation des électrons dans le systčme p crée un champ induit qui fait résoner les protons pour un champ plus faible ŕ l’extérieur du cône d’anisotropie, et pour un champ plus fort ŕ l’intérieur de ce cône. Donc les protons éthylčniques résonnent ŕ champ faible :
d = 5,64 ppm pour l’éthčne.

zone A : intérieur du cône d’anisotropie, déplacement de la résonance vers les champs forts .
zone B : déplacement vers les champs faibles.
Exemple du propčne : massif trčs complexe entre 4,9 et 5,3
ppm 
L’étude de l’hydrogčnation de divers alcčnes isomčres en un alcane identique permet de comparer les stabilités des doubles liaisons en fonction de leurs substituants.
Exemple : comparaison du but-2-čne Z et du but-2-čne E
![]() = enthalpie
d’hydrogčnation du but-2-čne Z
= enthalpie
d’hydrogčnation du but-2-čne Z
![]() = enthalpie
d’hydrogčnation du but-2-čne E
= enthalpie
d’hydrogčnation du but-2-čne E
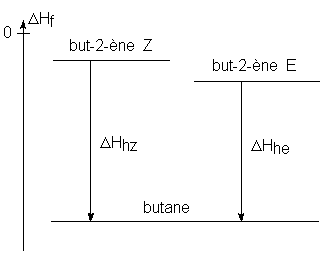
Cette différence de stabilité provient des contraintes stériques, plus importantes dans le Z que dans le E.
Autres exemples :
– les doubles liaisons exocycliques sont moins stables :
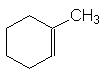
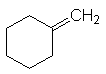
![]()
![]()
![]()
– la substitution par des groupes donneurs augmente la stabilité de la double liaison :
![]()
3.5. Hydrogčnation catalytique.
L’hydrogénation des alcčnes est exothermique (cf 3.4.), mais son énergie d’activation est élevée et elle nécessite un catalyseur pour que sa vitesse soit significative. Le diagramme énergie / avancement est le suivant pour cette réaction :
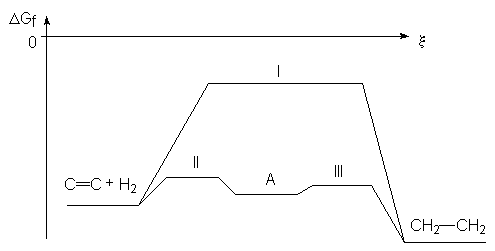
I : état de transition du type ![]()
II : état de transition du type ![]()
oů M est un atome du métal catalyseur
(on a a aussi ŕ ce moment lŕ) ![]()
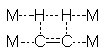 III : état de transition du type ci-contre oů les atomes du métal
permettent, grâce ŕ leur disposition, le rapprochement des atomes de C et de H.
III : état de transition du type ci-contre oů les atomes du métal
permettent, grâce ŕ leur disposition, le rapprochement des atomes de C et de H.
Le métal le plus utilisé est le Nickel. Le palladium peut également permettre l’hydrogčnation les alcčnes trčs réactifs. Le nickel nécessite une température de 25°C et une pression de 50 bars en hydrogčne. Par contre, le palladium et le platine permettent d’hydrogčner sous une pression de 1 ŕ 4 bars. Comme on le voit ci-dessus, l’addition est syncoplanaire. Ainsi, le 3,4-diméthyl-hex-3-čne Z sera-t-il hydrogčné en 3,4-diméthyl hexane méso.
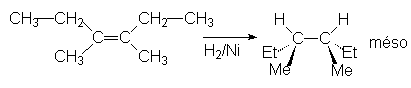
On peut aussi utiliser la diimide, formée in situ :
![]()
4. Addition électrophile sur la double liaison.
4.1. Mécanisme.
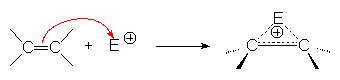
Cet ion " onium " existe sous deux formes mésomčres :
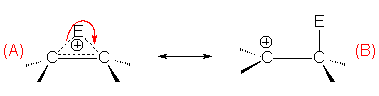
La premičre forme permet d’expliquer la stéréochimie de l’addition électrophile, la seconde son orientation.
4.1.1. Stéréochimie de l’addition.
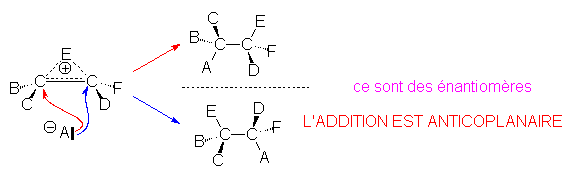
L’ion " onium " (B) ne peut ętre attaqué par le nucléophile A– associé ŕ l’électrophile qu’ŕ l’opposé du cycle trigonal. L’un ou l’autre carbone peut ętre attaqué.
Cette stéréospécificité est cependant relative. Elle dépend de la stabilité de l'ion "onium" intermédiaire. Celle-ci est importante avec un atome E gros et polarisable comme le brome, partielle avec le chlore, inexistante avec l'hydrogčne. C'est pourquoi cette stéréospécificité s'applique lors de l'addition de Br2 ou de BrOH, pour lesquels l'électrophile est Br+ , alors que l'on n'en tient pas compte lors de l'addition de composés ŕ hydrogčne mobile tels que l'eau.
4.1.2. Orientation de l’addition.
Celle-ci dépend de la position du carbocation intermédiaire (B). Ce sera le carbocation le plus stable (état intermédiaire) qui conduira ŕ la vitesse de formation du produit final le plus rapide. Il nous faut donc étudier les carbocations.
4.2. Les ions carbéniums (ou carbocations).
4.2.1. Structure.
Ils sont hybridés sp2 , avec une orbitale vacante pz
Ils sont stabilisés par les effets + I :
|
727 |
811 |
941 |
1078 |
 |
 |
|
 |
ou par les effets + E (résonance ) :
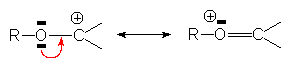
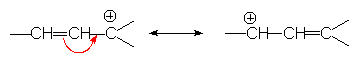
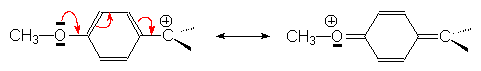
Certains carbocations sont trčs instables et se réarragent immédiatement par attaque électrophile intramoléculaire d’un doublet s , pour donner un carbocation plus stable (secondaire ou tertiaire): c’est le réarrangement de WAGNER - MEERWEIN.
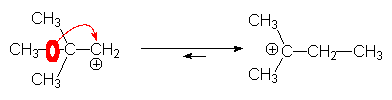
– départ d’un anion, ou d’azote :
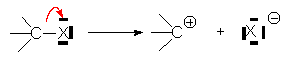
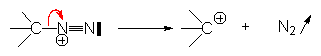
– départ d’un groupe électronégatif ŕ la suite de l’attaque de ce groupe par une structure électrophile :
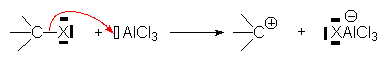
– attaque d’un doublet p :
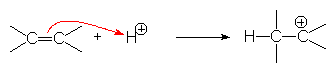
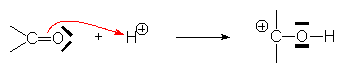
![]()
4.2.4. Orientation de l’addition électrophile sur les alcčnes.
L’addition d’un composé hétéroatomique (HCl par exemple) se fait donc de maničre ŕ ce que le carbocation intermédiaire obtenu soit le plus stable. C’est la rčgle de MARKOWNIKOW :
Lorsqu’un alcčne n’est substitué que par des groupes alkyle, l’addition électrophile se fait de maničre ŕ ce que le groupe électrophile se fixe sur le carbone le moins substitué :
![]()
Exception: lorsque l’un des carbones sp2 est substitué par des groupes attracteurs, l’électrophile se fixe sur ce carbone :
![]()
4.3. Vitesse de l’addition électrophile.
Celle-ci est d’autant plus rapide que l’état de transition correspondant ŕ la réaction lente (l’attaque de l’électrophile) est plus stable : il suffit de connaître l’énergie de formation du carbocation intermédiaire, proche de celle du premier état de transition. La vitesse est donc d’autant plus importante que les substituants sont donneurs. La stabilité des produits finaux étant semblable, la réaction sera surtout cinétique.
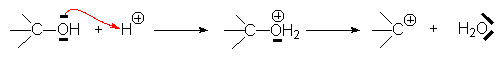
4.4.1. Addition d’acides.
![]()
Par exemple l’addition de HCl, HBr, CF3COOH
sur l’éthčne CH2 == CH2 donne CH3CH2Cl,
CH3CH2Br, et CH3CH2OCOCF3. Ces
acides libčrent facilement des protons. D’autres sont beaucoup moins forts et
libčrent peu de protons. Il faut alors ajouter des protons grâce ŕ un acide fort
présentant un anion peu nucléophile tel que ![]() . L’acide sulfurique ou l’acide paratolučnesulfonique
. L’acide sulfurique ou l’acide paratolučnesulfonique ![]() sont de trčs bonnes sources
de protons. Ainsi, l’on peut additionner des acides faibles présentant des anions
nucléophiles tels que H2O ou CH3COOH en présence de tels acides
forts :
sont de trčs bonnes sources
de protons. Ainsi, l’on peut additionner des acides faibles présentant des anions
nucléophiles tels que H2O ou CH3COOH en présence de tels acides
forts :
![]()
Pour obtenir le 2,2-diméthyl-propanol-2, dont le carbocation intermédiaire se forme trčs facilement, on utilise industriellement H2SO4 ŕ 10% ŕ 25°C, milieu suffisamment protonant pour hydrater le 2-méthyl-propčne :
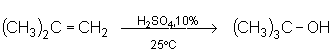
sous l’effet du champ créé par les électrons de
l’éthylčnique, les halogčnes sont polarisés et apparaît un cation électrophile
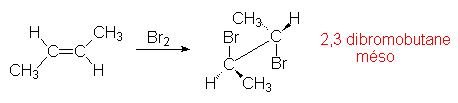
et un anion nucléophile : ![]() . L’addition, comme précédemment est anticoplanaire. La stéréochimie
des produits obtenus dépend de ce mécanisme :
. L’addition, comme précédemment est anticoplanaire. La stéréochimie
des produits obtenus dépend de ce mécanisme :
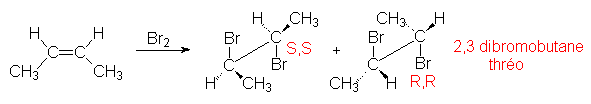
4.4.3. Formation d’halohydrines.
![]()
de męme avec ![]() ou
ou ![]()
L’addition est anticoplanaire (stéréospécificité stricte avec HOBr).
4.4.4. Dimérisation cationique.
Cette réaction est catalysée par les acides :
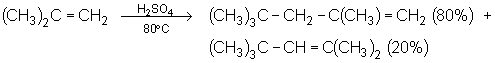
Le 2,4,4-pent-1-čne est plus stable ŕ cause de la répulsion entre les groupes tertiobutyl et méthyl du 2,4,4-pent-2-čne.
Voici le mécanisme: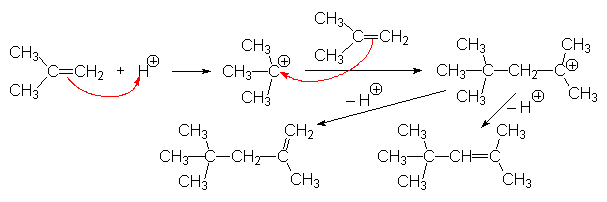
Si H2SO4 est trop concentré, on peut obtenir un polymčre, car H+ n’est plus arraché par l’eau.
5. Additions radicalaires sur les alcčnes.
En présence d’initiateurs radicalaires, les halogčnes et le bromure d’hydrogčne se décomposent en radicaux :
![]()
![]()
Lorsque l’on fait réagir HBr sur un alcčne en présence de peroxyde, le véritable électrophile est Brl : c’est le radical le plus stable et le plus réactif, contrairement ŕ Hl . On obtient une addition dont l’orientation est antimarkownikow : c’est l’effet KARASCH :
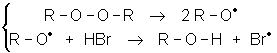 initiation
initiation
 propagation
propagation
Cette réaction se fait bien avec HBr, mais pas du tout avec HCl ni HI.
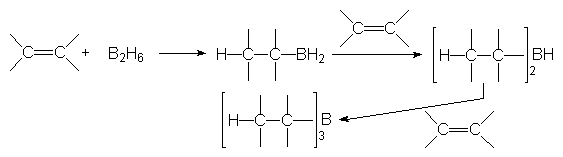
Cette réaction peut se faire avec les hydrures de bore ou les hydrures d’aluminium, mais principalement avec le diborane B2H6 :
On obtient des trialkylboranes qui sont des intermédiaires réactionnels trčs importants.
6.1. Orientation de l’addition.
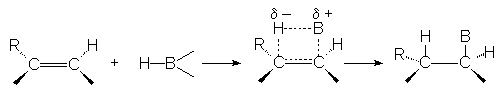 Cette addition est
" syncoplanaire " :
Cette addition est
" syncoplanaire " :
Cela ressemble ŕ l’addition de H2 en présence de catalyseur.
Le bore se fixe sur le carbone le moins substitué par des groupes donneurs, car B est moins électronégatif que H.
6.2. Réaction des alkylboranes.
6.2.1. Hydrolyse.
Celle-ci ne se fait bien qu’en milieu acide faible (CH3COOH + H2O). H2O attaque d’abord le bore, puis un H+ substitue du męme côté le bore :
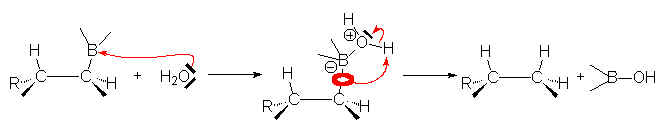
L’hydroboration suivie d’une hydrolyse acide conduit donc ŕ une hydrogčnation syncoplanaire d’un alcčne.
6.2.2. Hydrolyse oxydante par le peroxyde d’hydrogčne.
Cette réaction se fait en milieu basique. OH substitue B également du męme côté.
![]()
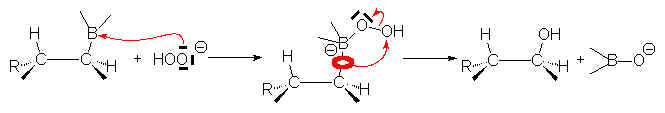
L’hydroboration suivie de l’action du peroxyde d’hydrogčne en milieu basique conduit ŕ une addition d’H2O syncoplanaire et antimarkownikow sur un alcčne.
6.2.3. Autres réactions des alkylboranes.
La réaction du sulfate d’hydroxylamine H2N — O — SO3H sur l’alkylborane conduit finalement ŕ l’addition syncoplanaire et antimarkownikow de NH3 sur l’alcčne :
![]()
De męme :
![]()
Et :
![]()
7. Autres additions
Ce procédé permet de synthétiser industriellement des
aldéhydes puis des alcools par addition de monoxyde de carbone et d’hydrogčne sur
un alcčne en présence d’un catalyseur qui est généralement
l’octocarbonylodicobalt ![]() :
:
![]()
Cet aldéhyde peut ensuite ętre réduit en butan-1-ol
Ils sont fortement électrophiles par leur lacune et attaquent les doublets p et s :
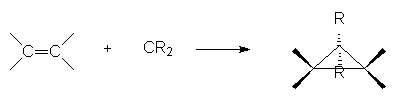
8. Réactions d’oxydation.
Industriellement, l’oxiranne (ou oxyde d’éthylčne, ou époxyde), est synthétisé par oxydation en présence d’argent :
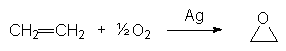
Au laboratoire, les peracides organiques oxydent les alcčnes en oxirannes :
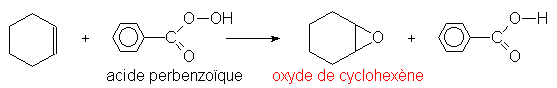
La vitesse de la réaction sera d’autant plus importante que la double liaison est substituée par des groupements donneurs d’électrons.
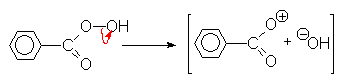
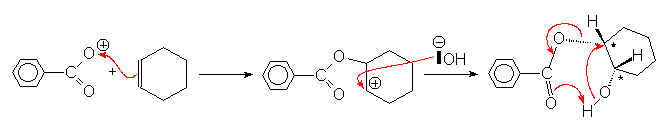
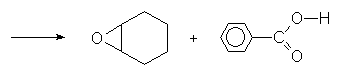
Mais ce mécanisme n’explique pas la rétention de configuration des carbones marqués *. Aussi est-il plus correct d’envisager un mécanisme totalement concerté.
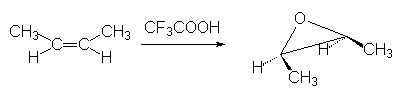
L’addition est donc cis : le but-2-čne Z donne ainsi le 2,3-diméthyl oxiranne méso :
Si on utilise un peracide fort (HCOOOH), l’époxyde formé intermédiairement est hydrolysé en anti-hydroxy-ester, car la base HCOO–
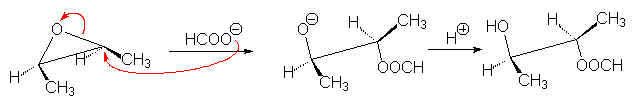
L’hydrolyse alcaline de cet a-hydroxy-ester donne un diol-1,2. Le résultat est une anti-bis-hydroxylation. On obtient le męme résultat en formant in situ l’acide perméthanoďque :
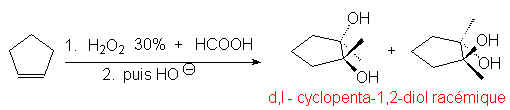
8.2. Syn (ou cis) hydroxylation.
Deux réactifs essentiels : ![]() , K+ et OsO4 (cher et
toxique)
, K+ et OsO4 (cher et
toxique)
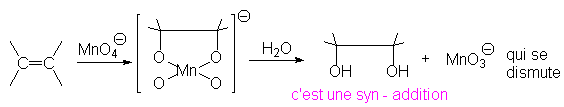
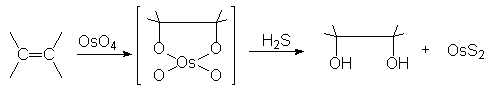
Ainsi le but-2-čne Z donnera-t-il le buta-2,3-diol méso par ce type de réaction.
O3 réagit ŕ basse température avec tous les alcčnes :
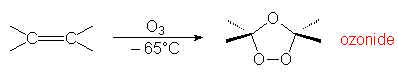
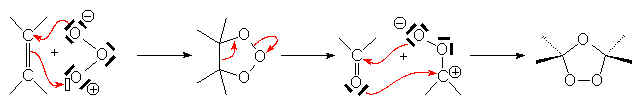
Les ozonides sont dangereux et explosifs. Ils sont ensuite traités par divers réducteurs pour donner des aldéhydes et cétones isolables et identifiables : Zn, CH3COOH ou H2/Pd , I– , PF3. Si on décompose l’ozonide par un excčs d’eau oxygčnée dans l’acide acétique, on obtient un acide, dans le cas oů le traitement réducteur donne un aldéhyde.
C’est l’oxydation catalysée de l’éthčne en
éthanal par l’oxygčne de l’air. Le catalyseur est le chlorure palladeux ![]()
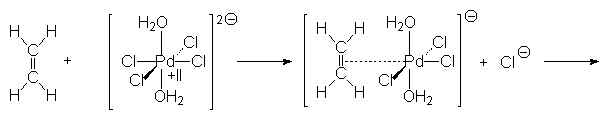
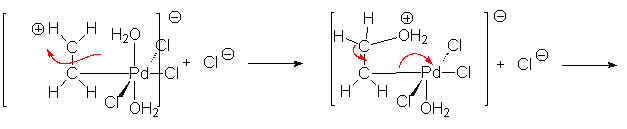
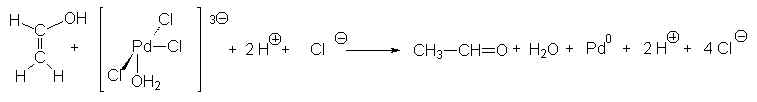
Puis Pd0 est oxydé par Cu2+
![]()
L’oxygčne de l’air oxyde ensuite Cu :
![]()
Globalement, la réaction s’écrit donc :
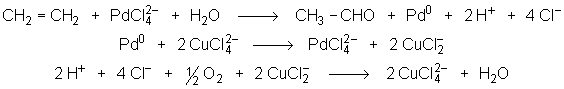
![]()
8.5. Oxydation par les halogčnes.
Oxydation allylique : ŕ haute température ( > ŕ 500°C), le chlore opčre une substitution radicalaire sur les hydrogčnes d'un carbone sp3 (tétračdrique) situé en a d'une double liaison Carbone – Carbone :
![]()
9. Les dičnes conjugués.
Ce sont des dičnes n, n+2. Leurs propriétés sont différentes de celles des dičnes isolés (n, n+m, avec m > 3) ou des dičnes cumulés tels les allčnes :
R – CH = C = CH – R’
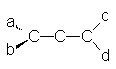
Le propadične en est un. Appelé aussi "allčne". La géométrie des liaisons est la suivante :
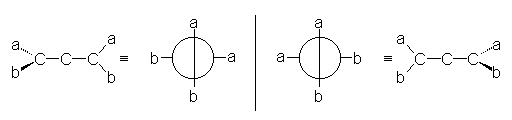
Les deux systčmes p étant perpendiculaires, les liaisons a et b sont dans un plan perpendiculaire au plan contenant les liaisons c et d. Cela entraîne l’isomérie "allčnique", qui est une isomčrie optique.
Pour qu’il y ait isomérie "allčnique, il suffit que les deux carbones terminaux ne portent pas de substituants identiques.
9.1.2. Dičnes conjugués: résonance.
Nous avons déjŕ vu comment la conjugaison augmentait la stabilité de ces molécules (cf énergie de résonance). Celle-ci peut ętre mise en évidence par la comparaison des enthalpies d’hydrogčnation du buta-1,3-dične s-trans (-238,7 kJ.mol–1) et du penta-1,4-dične (-254,1 kJ.mol–1). Le premier est conjugué, l’autre pas, tout en possčdant le męme nombre de doubles liaisons. L’énergie de résonance vaut donc 15,4 kJ.mol–1.
D’autre part, la nucléophilie de ces alcčnes augmente, car la plus haute orbitale occupée, qui contient le doublet nucléophile, a une énergie moins basse que dans le cas des alcčnes, donc ce doublet est plus disponible. La réactivité des dičnes conjugués est donc plus importante que celle des alcčnes.
9.2. Additions électrophiles conjuguées.
La vitesse de réaction est augmentée, car le carbocation intermédiaire est stabilisé par résonance avec la double liaison restante :
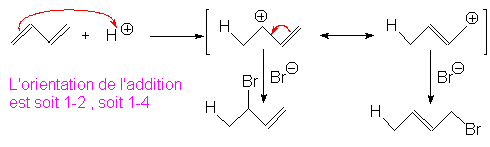
Les proportions relatives dépendent de la température, de la durée de la réaction et du solvant.
Les solvants polaires favorisent l’addition 1-4.
Un temps de réaction important crée des conditions propres ŕ un contrôle thermodynamique, nous allons voir que dans ces conditions, c’est l’addition 1-4 qui est favorisée.
Ŕ – 60°C, on obtient, pour la réaction précédente, 80% de 1-2 et 20% de 1-4.
Ŕ +25°C, on obtient 20% de 1-2 et 80% de 1-4.
Comme nous l’avons montré, le premier cas correspond ŕ un contrôle cinétique de la réaction, le second ŕ un contrôle thermodynamique :
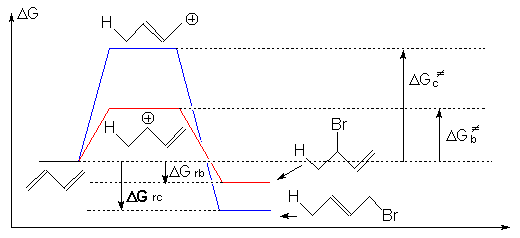
Le carbocation le plus stable est le carbocation secondaire. L’alcčne final le plus stable sera celui oů la double liaison est la plus substituée.
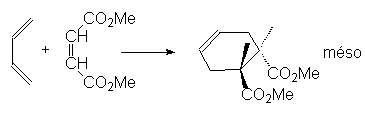
9.3. Cycloadditions. Réaction de Diels-Alder.
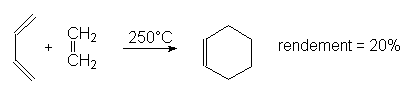
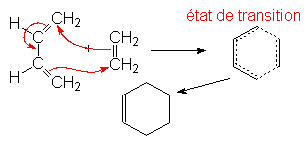
C’est une addition 1-4 d’un alcčne ŕ un dične conjugué : c’est une cycloaddition car elle donne un composé cyclique. Pour réagir, le dične doit ętre de conformation s-cis. La réaction est d’autant plus rapide que l’alcčne (ou philodične) est pauvre en électron (substitué par des groupes attracteurs d’électrons) et que le dične en est riche.
Exemple :
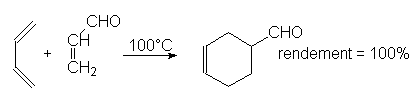
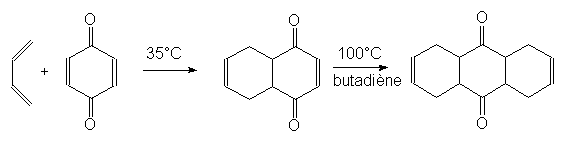
Les configurations sont conservées, donc les liaisons s se forment avant la rupture des p.
Quand le dične est cyclique, deux isomčres sont obtenus :
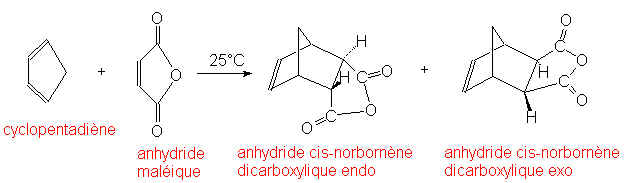
C’est l’état de transition qui donne l’adduct endo qui est le plus stable.
Ces réactions sont des réactions en chaîne. Elles sont catalysées par la plupart des entités instables: cations ou acides de Lewis, anions, radicaux. Les éthylčniques les plus réactifs sont ceux qui sont riches en électrons, donc substitués par des groupements donneurs. Par exemple:
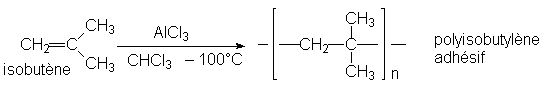
Les polymčres obtenus ŕ partir de dičnes peuvent se relier par concaténation. Par exemple, le copolymčre isobutčne-isoprčne donne par concaténation le caoutchouc butyle:
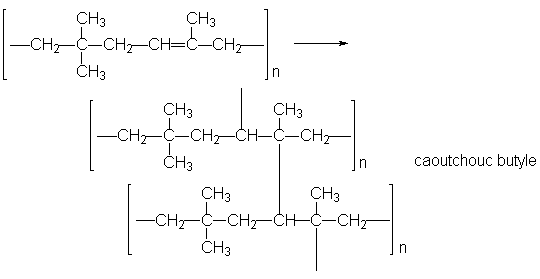
La polymérisation anionique se fait par exemple grâce aux
composés suivants : ![]() ,
Li.
,
Li.
L’isoprčne est ainsi polymérisé par Li en caoutchouc corail, presque entičrement cis (alors que le caoutchouc naturel est entičrement cis)
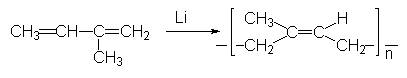
Les polymérisations radicalaires sont catalysées par les
peroxydes, tels le peroxyde de benzoyle ![]() . On obtient le polystyrčne :
. On obtient le polystyrčne :
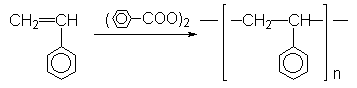
11. Méthodes de synthčse des alcčnes.
11.1. Déshydrohalogčnation des halogčnures d’alkyle.
![]()
11.2. Déshydratation des alcools.
![]()
11.3. Déshalogčnation des halogčnures vicinaux.
![]()
11.4. Pyrolyse d’hydroxydes d’ammonium quaternaires ( réaction d’Hoffmann).
![]()
11.5. Pyrolyse des esters.
![]()
11.6. Pyrolyse des oxydes d’amine.
![]()
11.7. Réaction de WITTIG.
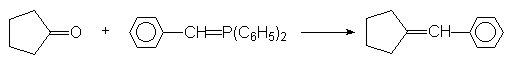
11.8. Réduction des acétyléniques.