1. Structure de la triple liaison.
1.1. Hybridation des carbones.
Les deux carbones concernés sont hybridés sp. Cela entraîne une électronégativité de 3,1 dans léchelle de PAULING. Ces hybrides sp (2 par carbone, formant entre eux un angle de 180°), vont permettre la création des liaisons s , avec les atomes voisins et entre eux. La liaison s Csp Csp a une énergie de 470 kJ.mol1. Elle est donc très stable et courte (0,12 nm). Sur chaque atome de carbone, il reste deux OA non hybridées parallèles deux à deux. Elles vont se recouvrir deux à deux pour donner deux OM p (pz et px par exemple) et deux OM p* (pz* et px*). Les pz et px sont perpendiculaires. Les deux liaisons p sont beaucoup moins stables que la s (175 kJ.mol1). Au total, lénergie de la liaison triple C º C vaut 820 kJ.mol1.
Contrairement à la double liaison, les électrons, sous leffet de H extérieur, circulent dans les OM p à la manière dun solénoïde dont laxe serait la liaison triple, le cône danisotropie contient donc les éventuels protons portés par le carbone sp, si bien que ces protons ont un déplacement chimique plus faible que celui attendu, compte tenu de lélectronégativité importante du carbone sp. Le déplacement chimique du proton acétylénique est compris entre 2,4 et 2,7 ppm.
Un monoalcyne est dénommé en remplaçant le suffixe ane par yne : CH3 CH2 C º CH : but-1-yne.
2. Propriétés physicochimiques.
2.1. Températures de changement détat.
Ces hydrocarbures sont très effilés, les forces de Van der Waals sont donc importantes, et les températures de changement détat plus élevées que celles des alcanes homologues : teb C2H4 = 102°C teb C2H2 = 83°C
Léthyne (ou acétylène) est obtenu industriellement par réduction du carbonate de calcium par le coke à haute température (vers 1500°C) :
CaCO3 + 3 C
® CO + CaC2 (carbure de calcium,
structure de type NaCl, lanion étant: ![]() ). Celui-ci shydrolyse très
facilement (lampes à acétylène) en éthyne :
). Celui-ci shydrolyse très
facilement (lampes à acétylène) en éthyne :
CaC2 + H2O ® Ca(OH)2 + HC º CH
Lenthalpie de combustion de C2H2 est très élevée, la flamme oxyacétylénique pouvant atteindre une température de 2900°C. Aussi la manipulation de léthyne est-elle hasardeuse à létat sec. Lodeur caractéristique de lacétylène obtenu par hydrolyse provient de lhydrolyse concommittante du phosphure de calcium, produit parasite de la réaction de CaCO3 avec le carbone contenant du phosphore :
Ca3P2 + H2O ® Ca(OH)2 + PH3 (phosphine nauséabond)
La forte électronégativité du Csp stabilise les charges négatives (plus que ne le fait lazote par exemple). Le pKA du couple acide/base RC º C H / RC º C vaut 25. Lamidure de sodium transformera donc totalement lalcyne en alcynure. Lhydroxyde de sodium ne donne lieu quà un équilibre partiel (RC º CH + OH ® RC º C + H2O ). Cet équilibre peut être déplacé vers la droite en précipitant lanion alcynure avec des cations tels que Cu+ ou Ag+ . On obtient des composés insolubles dans leau et de diverses couleurs : CuC º CCu est rouge, RC º CCu est jaune, RC º CAg est blanc. Tous ces composés sont explosifs à sec.
3. Addition électrophile aux alcynes.
Les règles dorientation et de stéréochimie appliquées aux alcènes sappliquent également aux alcynes. Ainsi :
CH3 C º C CH3 + Br2 ¾ ® CH3 CBr = CBr CH3 (E)
Laddition est plus rapide quavec les alcènes, vu la faiblesse des liaisons p , et plus exothermique, ce qui entraîne parfois une réaction destructrice : le chlore (et le fluor bien sûr) enflamme léthyne.
Lalcyne peut additionner deux moles dhalogènure dhydrogène
![]()
Lorientation de la deuxième addition est liée aux effets mésomères donneurs du premier brome.
Laddition deau et dacide acétique, acides trop faibles, nécessite un catalyseur : Hg2+ + H2SO4 :
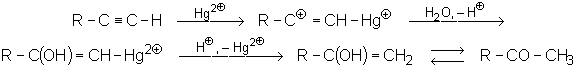
On obtient toujours le carbonyle le plus substitué.
Autre exemple : ![]()
(acétate de vinyle, utilisé pour former des polyacétates de vinyle, matières plastiques).
Laddition de B2H6 suit les mêmes règles dorientation et de stéréochimie que pour les alcènes. Suivie dune réaction avec le peroxyde dhydrogène H2O2 en milieu basique, on obtient une addition deau antimarkownikow et syn. Par exemple :
![]()
4.1. Hydrogènation catalytique.
 En
présence de Nickel Raney, les deux insaturations sont hydrogènées. Pour
sarrêter aux alcènes, il faut utiliser un catalyseur désactivé : Pour cela, on
utilise du palladium pulvérisé avec du sulfate de baryum, le tout imbibé de
quinoléine :
En
présence de Nickel Raney, les deux insaturations sont hydrogènées. Pour
sarrêter aux alcènes, il faut utiliser un catalyseur désactivé : Pour cela, on
utilise du palladium pulvérisé avec du sulfate de baryum, le tout imbibé de
quinoléine :
Laddition est syn.
![]()
Suivie dhydrolyse en milieu acétique, elle conduit également à une hydrogènation syn.
Elle est obtenue par action du sodium dans lammoniac liquide :
![]()
Lozonolyse suivie dune hydrolyse réductrice (CH3COOH , Zn) donne deux acides :
![]()
Loxydation par K2Cr2O7 ou KMnO4 donne la même chose.
Les alcynes substitués par des groupements attracteurs sont de bons diènophiles :
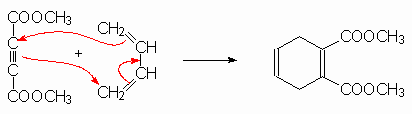
Par contre lalcyne est un mauvais diène, car il
ne peut se positionner en conformation s-cis. La réaction nécessite une haute
température: ![]() est un mauvais diène.
est un mauvais diène.
La base conjuguée de lalcyne vrai est une base forte et donc un bon nucléophile dans les solvants aprotiques et les solvants protiques peu polaires.
Cet alcynure peut donner des réactions de substitution nucléophile et daddition nucléophile.
Les dérivés halogénés (primaires de préférence), réagissent selon une SN2 (voir chapitre suivant)
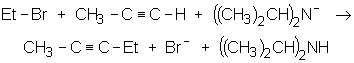
Toujours en présence de base forte, les alcynes sadditionnent aux dérivés carbonylés :
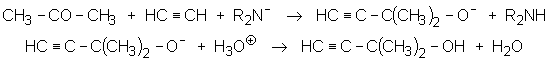
« méthylbutinol »
Traités par NaNH2 , les alcynes disubstitués se transforment en alcynes vrais :
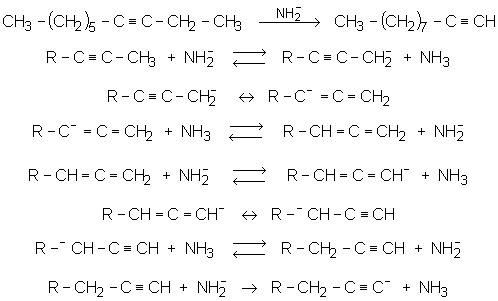
La dernière réaction nest pas un équilibre, cest pour cela que la réaction globale est déplacée vers la droite.
En présence de méthylate de sodium MeONa , les alcynes vrais sont eux transformés en alcynes disubstitués. Ces deux réactions demandent une durée très longue (plusieurs heures, voire plusieurs jours).