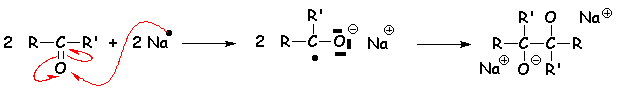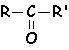 ou R et R'
représentent des groupements alkyls, alkènyls ou alkynyls (R-Cº C-), ou un atome d'hydrogène.
ou R et R'
représentent des groupements alkyls, alkènyls ou alkynyls (R-Cº C-), ou un atome d'hydrogène.1. Structure. Propriétés physiques.
Ces composés sont de la forme : ou R et R'
représentent des groupements alkyls, alkènyls ou alkynyls (R-Cº C-), ou un atome d'hydrogène.
Lorsque R ou R' est un H, ce dérivé carbonylé est un aldéhyde. Cet hydrogène confère des propriétés réductrices aux aldéhydes. Lorsque R et R' sont tous deux des groupements alkyls, alkènyls ou alkynyls, le dérivé carbonylé est une cétone.
Les dérivés carbonylés sont des composés polaires, mais ne présentent pas de liaisons intermoléculaires fortes comme les liaisons hydrogène :
moment dipolaire liaison H Teb alcools oui oui 90°C dérivé carbonylé oui non 55°C alcane non non - 100°C
NB : nous ne parlons ici que des interactions attractives entre molécules polaires (interactions dipôle-dipôle ou forces de Keesom)
2. Réactivité des dérivés carbonylés
trois sites particuliers : Le
carbone du carbonyle présente un charge partielle positive. Il
sera donc sensible aux attaques de nucléophiles. Les doublets de
L'oxygène sont basiques et pourront donc être facilement protonés.
L'hydrogène en a du carbonyle est
acide.
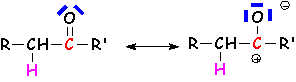
Il s 'agit de la fixation d'une structure nucléophile sur le carbone, avec fixation de L'électrophile associé (souvent H+) sur L'oxygène. Deux mécanismes essentiels :
Le nucléophile est assez puissant pour attaquer le premier. Cette réaction se fait dans un milieu qui préserve L'intégrité de ce nucléophile, donc un milieu basique. La protonation se fait généralement lors de L'hydrolyse du composé d'addition obtenu :
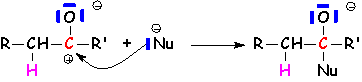
La réaction se fait en présence de protons libres (acides forts) ou d'acides faibles pouvant créer des liaisons hydrogène, tels que les acides carboxyliques (il s 'agit alors de la catalyse acide généralisée) :
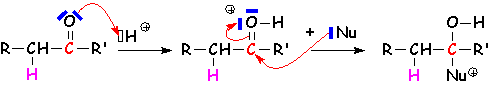
![]() se stabilise généralement en perdant un de ses
protons.
se stabilise généralement en perdant un de ses
protons.
2.1. Mobilité des protons en a du carbonyle
Il s 'agit véritablement ici d'étudier L'action de la base sur le dérivé carbonylé :
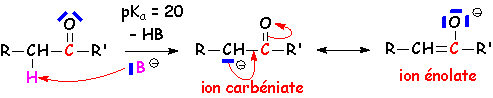
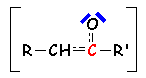
Ces deux formes anioniques représentent la même structure (d'où la relative stabilité de cette structure). L'hybride de résonance correspondant est :
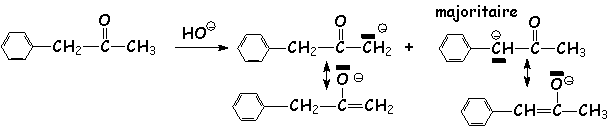
Régiosélectivité de la formation de L'énolate : En effet, dans le cas d'une cétone dissymétrique, quel est L'énolate majoritaire ? Dans le cas d'une réaction où l'équilibre n'est pas atteint(contrôle cinétique en présence de bases faibles telles que OH- ou EtO-)(voir l'aldolisation), on obtient majoritairement le carbéniate le plus stable, c.a.d. le moins substitué par des groupements donneurs :
Par contre, Si L'anion du carbéniate est conjugué, il sera majoritaire :
Selon la force de la base, la formation de L'énolate sera plus
ou moins complète : en milieu HO -, la constante
d'équilibre vaut 10 -5 environ, en milieu ![]() , elle
vaut 1010. Dans ce dernier cas, la forme "énolate (avec la double
liaison)" implique une plus grande stabilité de l'énolate le plus substitué
(voir l'alkylation des énolates)
, elle
vaut 1010. Dans ce dernier cas, la forme "énolate (avec la double
liaison)" implique une plus grande stabilité de l'énolate le plus substitué
(voir l'alkylation des énolates)
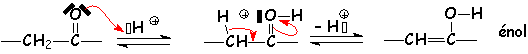
Le problème de la régiosélectivité se pose également ici : L'énol qui se forme est celui qui possède la double liaison C = C la plus stable. Ainsi, dans L'exemple précédent, le composé majoritaire est :
Les énols les plus stables sont donc ceux qui dérivent des cétones.
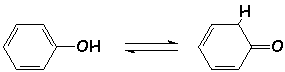
Les phénols sont des énols très stables (rôle de L'aromaticité) :
Cet équilibre s 'appelle L'équilibre (ou tautomérie) cétoénolique. On le nomme plus généralement prototropie (déplacement équilibré d'un proton).
Autre exemple de prototropie : équilibre C-hydroxyimine - amide.
3. Additions nucléophiles sur les dérivés carbonylés.
3.1. Catalyse basique.
Cette addition est généralement suivie d'une hydrolyse en milieu acide, ce qui permet de protoner L'ion O -.
Les cétones réagissent moins bien que les aldéhydes vis-à-vis de L'addition nucléophile,
Les carbanions les plus faciles à obtenir sont les organométalliques, dont font également partie les ions alcynure. Nous verrons plus loin que les carbéniates peuvent également s 'additionner (§ 4)
Exemples : ![]()
Nous avons déjà vu les organolithiens (qui sont les plus ionisés) :
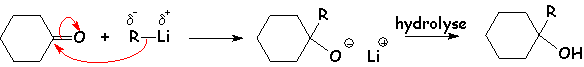
Dans le cas des organomagnésiens, le mécanisme réel est plus complexe et met en jeu deux molécules d'organomagnésiens pour une de dérivé carbonylé :
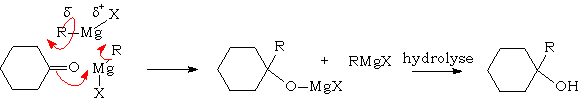
Lorsque les molécules sont encombrées, le comportement des divers organométalliques diffèrent : les organolithiens réagissent selon la norme indiquée plus haut, alors que les organomagnésiens donneront généralement une réduction des dérivés carbonylés encombrés :
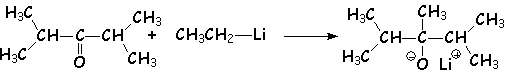
Mais :
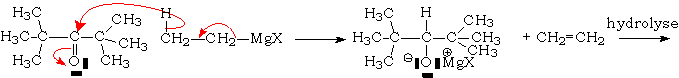
La réaction d'addition nucléophile des organométalliques sur les dérivés carbonylés a,b-éthyléniques, diffère aussi selon la nature de cet organométallique :
- Les lithiens donnent toujours une addition 1,2 :

- En fonction de L'encombrement de la position 4, les organomagnésien donneront tantôt une addition 1,2 (pos. 4 encombrée), 1,4 (pos. 4 non encombrée), ou un mélange des deux dans le cas intermédiaire (CH3 n 'est pas très encombrant) :
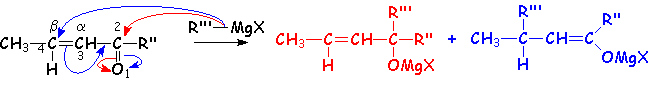
L'hydrolyse libère un énol dans le second cas, qui se tautomérise :
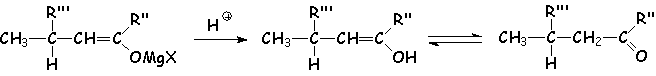
- Pour que L'addition soit uniquement 1,4 " L'encombrement du C4, on a recours aux organocuprates, de formule R2CuLi :
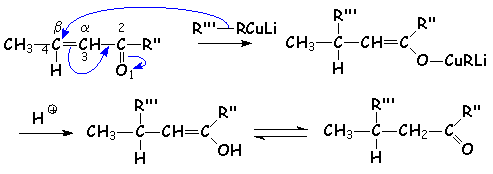
: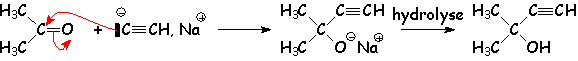
Ces ions alcynures peuvent être obtenus soit par action de NaNH2, soit par action d'un organomagnésien plus basique :
![]()
Il s 'additionne sur le carbonyle :
L'étude de cette réaction va nous permettre de dégager quelques règles générales concernant L'addition nucléophile (ces règles pourront être appliquées dans la suite du cours) :
Le dérivé carbonylé sera d'autant plus sensible à L'addition nucléophile que le carbone fonctionnel est pauvre en électron : donc les aldéhydes, qui subissent moins d'effets donneurs que les cétones (voir les effets électroniques des groupes alkyles), seront plus réactifs qu 'elles. Le méthanal est le plus sensible de tous. Si le carbonyle est substitué par des groupements à effet attracteur, ils seront encore plus réactifs.
d'autre part, la diminution de L'angle de liaison lors de la réaction d'addition (de 120° à 109°) va augmenter la contrainte d'autant plus que les groupements fixés sur le carbonyle sont gros. Là encore, c 'est un argument qui va dans le sens d'une plus grande stabilité des produits d'addition obtenus à partir des aldéhydes :
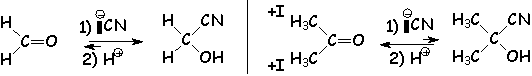
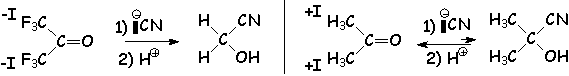
Condensation benzoïne
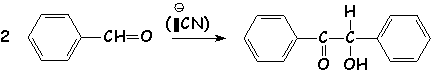
Ne se produit qu 'avec les aldéhydes aromatiques, car le substituant du carbonyle (ici le cycle aromatique), est capable de stabiliser les anions :
Le mécanisme de cette condensation est le suivant :
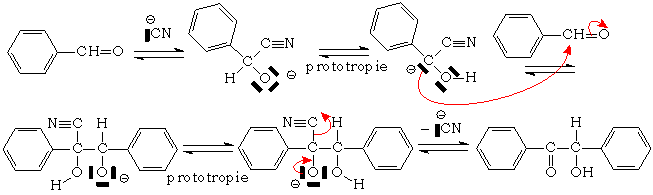
Cette réaction est à contrôle thermodynamique. Il s 'agit d'un équilibre qui est déplacé vers la formation du composé le plus stable qui est la benzoïne.
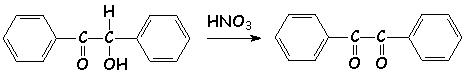
La benzoïne est facilement oxydée par L'acide nitrique concentré en benzile :
Le benzile subit ne milieu basique la transposition " benzilique ", qui est une transposition anionique, contrairement à toutes celles que nous avons vu jusqu 'à présent (Wagner-Meerwein, pinacolique, &), c 'est-à-dire qu 'elle est initiée par la présence d'un anion :
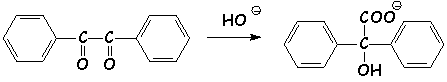
Voici son mécanisme :
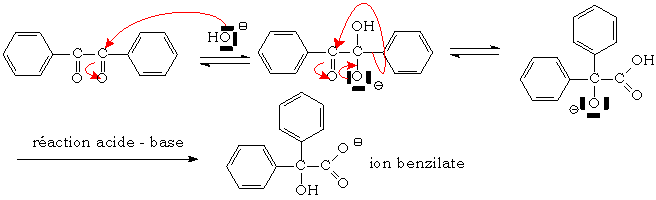
Cet ion benzilate est ensuite protoné en acide
benzilique :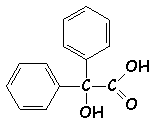
3.1.2. Addition d'ions hydrure : réactions de réduction.
On trouve L'ion hydrure sous la
forme d'hydrure de sodium NaH, qui est un composé ionique de la forme ![]() . Cet anion est un
nucléophile moyen, mais c 'est aussi une base très forte (anion petit et très
chargé). Sa basicité va le rendre inefficace pour qu 'il puisse s 'additionner
au groupement carbonyle (en effet, il arrachera plus facilement les H en a de
CO). Son caractère basique s 'estompe et son caractère nucléophile augmente
quand il sert de ligand dans certains complexes de L'aluminium ou du bore, qui
sont beaucoup plus volumineux : la charge négative n 'est plus concentrée
en un seul endroit de L'espace . Ce sont le tétrahydruroborate de sodium
(NaBH4) et le tétrahydruroaluminate de lithium (LiAlH4)
bien plus réactif :
. Cet anion est un
nucléophile moyen, mais c 'est aussi une base très forte (anion petit et très
chargé). Sa basicité va le rendre inefficace pour qu 'il puisse s 'additionner
au groupement carbonyle (en effet, il arrachera plus facilement les H en a de
CO). Son caractère basique s 'estompe et son caractère nucléophile augmente
quand il sert de ligand dans certains complexes de L'aluminium ou du bore, qui
sont beaucoup plus volumineux : la charge négative n 'est plus concentrée
en un seul endroit de L'espace . Ce sont le tétrahydruroborate de sodium
(NaBH4) et le tétrahydruroaluminate de lithium (LiAlH4)
bien plus réactif :
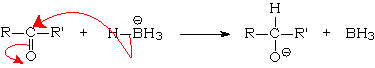
L'hydrolyse conduit à L'alcool.
Les acides et dérivés d'acide ne sont pas réduits par NaBH4, mais le sont par LiAlH4. Le premier est donc le réactif de choix pour la réduction exclusive des fonctions carbonyles.
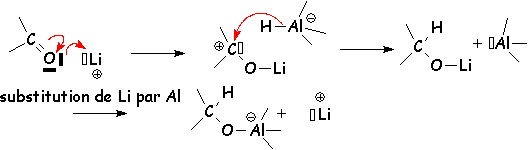
NB : (hors programme : rôle du cation ou contre-ion associé à l'aluminate )
Lors de la réduction par LiAlH4 le cation Li+ est indispensable pour activer le carbonyle :
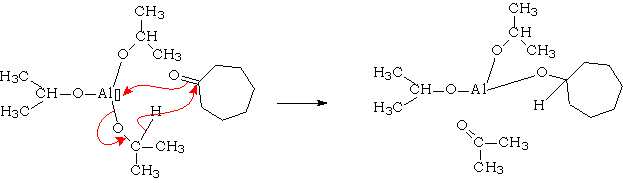
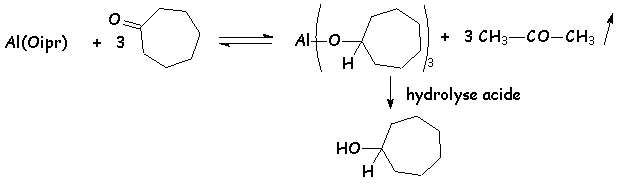
L'isopropylate d'aluminium est un donneur d'ion hydrure peu aggressif. Il servira à réduire des cétones " fragiles ". La réaction est un équilibre entre alcoolates et cétones, L'équilibre étant déplacé par distillation du composé le plus volatil qui est la propanone :
Le bilan est le suivant :
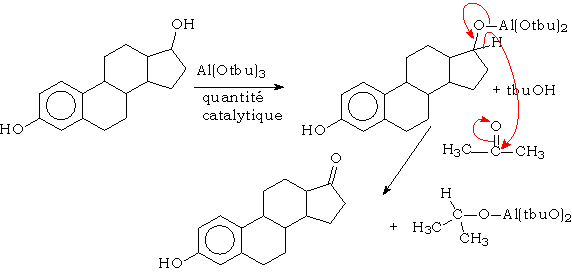
On peut également, par une réaction semblable, oxyder les alcools fragiles en cétones ou aldéhydes. Cette réaction, inverse de la précédente, s 'appelle la réaction d'Oppenauer :
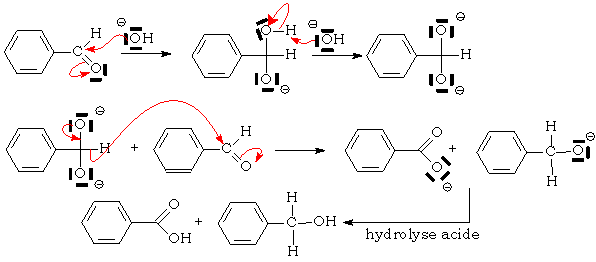
Cette réaction se produit en milieu OH - lorsque le carbone en a du carbonyle ne porte pas d'hydrogène : le benzaldéhyde, le glyoxal :
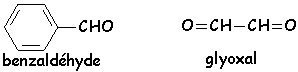
OH - ne peut jouer son rôle de base (arrache normalement un H en a du CO pour donner un énolate) et se comporte en nucléophile :
Le furfural est un autre exemple de composé pouvant subir la
réaction de Cannizzaro : 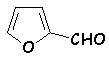
3.1.4. Combinaison bisulfitique : addition de composés soufrés.
L'ion hydrogénosulfite, possédant un doublet sur le carbone, s 'additionne aux aldéhydes, plus réactifs et moins encombrés, pour donner un ion a-hydroxysulfonate :
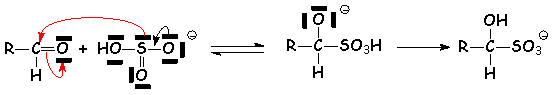
Cet a-hydroxysulfonate est soluble dans L'eau, ce qui n 'est généralement pas le cas de L'aldéhyde de départ. On pourra donc se servir de cette méthode pour séparer L'aldéhyde d'autres composés insolubles dans L'eau gênants. La solution aqueuse purifiée est alors traitée en milieu acide, ce qui régénère L'aldéhyde de départ, insoluble à nouveau dans L'eau.
Il s 'agit de la réaction d'un ylure de phosphore sur un dérivé
carbonylé (exemple : le méthylure de triphénylphosphonium :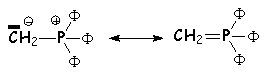 . Il s 'agit donc bien, au départ d'une attaque nucléophile
directe sur le carbonyle. Grâce aux orbitales d du phosphore, L'ylure est
stabilisé et on peut en représenter une autre forme mésomère.
. Il s 'agit donc bien, au départ d'une attaque nucléophile
directe sur le carbonyle. Grâce aux orbitales d du phosphore, L'ylure est
stabilisé et on peut en représenter une autre forme mésomère.
Synthèse de L'ylure :
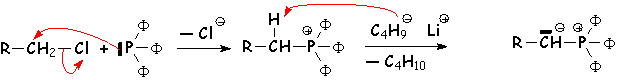
La triphénylphosphine, nucléophile, substitue(SN2) L'halogène d'un dérivé halogéné, puis une base très forte arrache un H en a de P pour donner L'ylure :
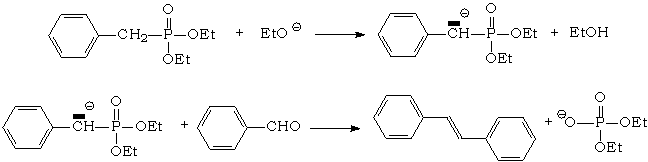
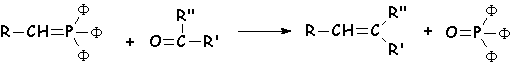 : Réaction de Wittig
: Réaction de Wittig
Les alcènes obtenus sont " généralement E " (les moins encombrés)
Réactions apparentées : Staudinger, Corey - Winter, Mitsunobu, Horner Wadsworth - Emmons
Par exemple, une réaction d'Arbuzov permet de former un alkylphosphonate :
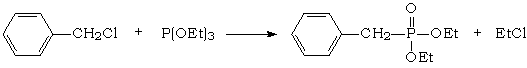
Puis l'action du benzaldéhyde sur l'anion obtenu par action d'une base forte sur l'alkylphosphonate forme un alcène E : C'est la réaction de Horner Wadsworth - Emmons :
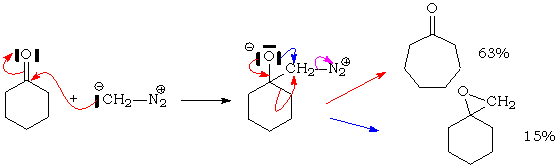
Le diazométhane donne généralement des "augmentations" de taille de cycle. Il y a insertion de CH2 à côté du C=O.
Le méthylure de diméthylsulfonium donne un oxiranne :
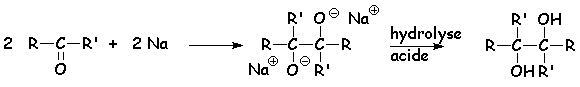
Les métaux fortement réducteurs peuvent, en milieu aprotique, dupliquer simultanément la cétone :
Dans le cas de la propanone, on obtient le pinacol. Le solvant aprotique qui convient est le benzène ou le toluène.
Le mécanisme de la duplication est le suivant :
Le zinc en milieu acide réduit les dérivés carbonylés en alcane (voir CLEMMENSEN)
Les acides forts (tels que H2SO4) ou faibles (CH3COOH) peuvent catalyser ces réactions d'addition nucléophile. En fait, il suffit que L'établissement d'une liaison hydrogène entre L'acide (ou H+) et L'oxygène du carbonyle augmente la charge partielle positive du carbone du carbonyle, ce qui le rendra bien plus sensible aux attaques nucléophiles :
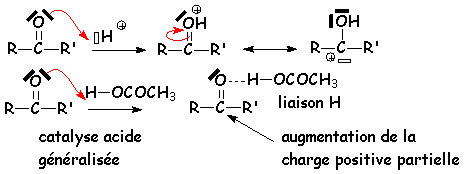
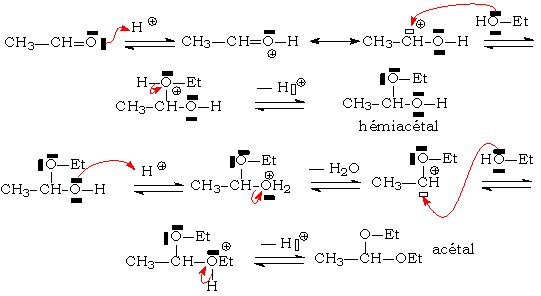
Les aldéhydes s'additionnent sur les monoalcools à pH ~ 1 pour donner un hémiacétal instable qui peut ensuite se condenser avec une autre molécule d'alcool pour donner un acétal :
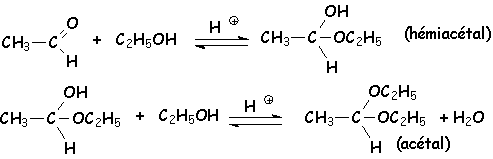
On déplace L'équilibre grâce à un dyn stark (le moyen de chauffage n'est pas figuré) :
.gif)
L'eau est éliminée car elle est non miscible et plus dense que L'azéotrope ternaire
Les cétones ne donnent pas les composés correspondants aux hémiacétals (hémicétals) et aux acétals (cétals), car la constante de L'équilibre est bien plus faible. Par contre, la création de cétals cycliques est possible, la structure étant stabilisée par la présence du cycle. Ainsi, les diols terminaux (éthane-1,2-diol, propane-1,3-diol, etc &) donnent-ils avec les cétones des 1,3-dioxa-cyclopentane et 1,3-dioxa-cyclohexane :
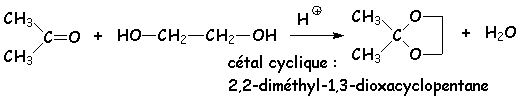
Mécanisme complet (pour un aldéhyde, valable pour une cétone) :
Cette réaction ne se fait qu'avec les dérivés carbonylés. (les esters par exemple ne peuvent donner ainsi des orthoesters :
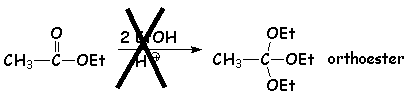
L'intérêt de la formation de ces acétals et cétals, c'est leur stabilité en milieu basique (base alcaline, ou base plus forte, tel qu'un organométallique). On pourra ainsi protéger un groupement carbonyle, si on doit faire réagir un autre groupement de la molécule en milieu basique (généralement, les bases provoquent L'aldolisation du dérivé carbonylé, ce que L'on veut éviter. Exemple :
NB : on trouve des hémiacétals stables en série cyclique ( à 5 ou 6 chaînons). L'exemple le plus important est celui des formes hémiacétaliques des oses. Par exemple, le glucose (b-D-glucopyranose) :
.gif)
Les thiols RSH réagissent en milieu acide avec tous les dérivés carbonylés pour donner des thioacétals (avec les aldéhydes) et des thiocétals (avec les cétones) :
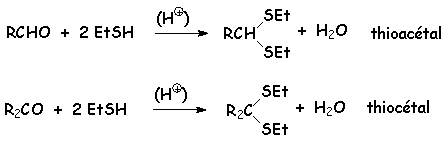
Récapitulation :
|
alcools |
thiols |
diols | |
|
Aldéhydes |
acétals |
thioacétals |
Acétals cycliques |
|
Cétones |
/ |
thiocétals |
Cétals cycliques |
Pour déprotéger les thiocétals, il faut les traiter en milieu acide par des sels mercuriques (les ions mercuriques donnent des liaisons covalentes très stables avec les anions soufrés) :
.gif)
Outre leur utilisation identique à celle des acétals (protection du groupement carbonyle), les thioacétals et thiocétals permettent de transformer un dérivé carbonylé en alcane :
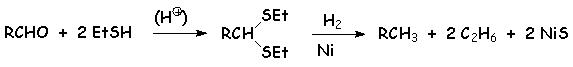
L'addition d'eau conduit à un diol géminé, selon :
.gif)
Cet équilibre est généralement très peu déplacé vers la droite, sauf dans les cas suivants :
Exemples : L'éthanal se trouve en solution aqueuse sous les deux formes : 50% sous la forme aldéhyde, 50% sous la forme diol géminé, alors qu 'en solution aqueuse, la propanone est uniquement sous la forme cétonique.
Par contre, le méthanal est totalement hydraté (), ainsi que le chloral :
![]()
3.2.4. Addition nucléophile de dérivés azotés G - NH2
Il s 'agit la plupart du temps d'une addition - élimination :
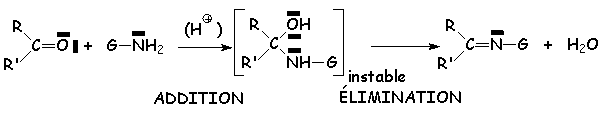
Cette réaction nécessite un pH compris entre 3 et 5, pour qu 'il y ait suffisamment de protons pour protoner L'oxygène du carbonyle, et pas trop pour éviter de protoner L'azote de G - NH2, ce qui supprimerait ses propriétés nucléophiles. Le mécanisme est le suivant :
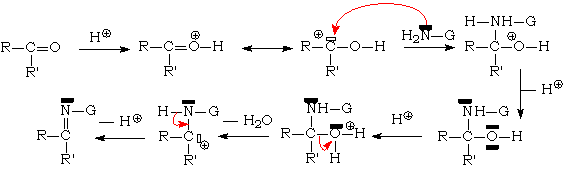
Le composé azoté est donc L'ammoniac ou une amine. L'amino-alcool intermédiaire est instable (sauf dans le cas de l'addition d'ammoniac sur l'éthanal), et le composé final s 'appelle une imine (aldimine lorsque L'on part d'un aldéhyde, cétimine lorsque L'on part d'une cétone. Le nom de ces composés est obtenu en considérant L'imine comme substituée sur L'azote par un groupement alkyle. Par exemple, L'imine obtenue par condensation de la méthylamine sur le propanal, s 'appelle la N-méthylpropan-1-imine : CH3CH2CH = N - CH3. Elles sont généralement instables et se polymérisent, en trimères cycliques ou en polymères à longue chaîne.
Le méthanal donne l'hexaméthylène tétramine avec l'ammoniac :
.gif)
Les imines aromatiques (obtenues à partir de benzaldéhyde et d'aniline par exemple) Sont plus stables et sont appelées " bases de Schiff ".
3.2.4.2. G º OH (condensation de L'hydroxylamine NH2OH)
On obtient une oxime, qui est un dérivé cristallisé des dérivés carbonylés, dont le point de fusion est très reproductible, et donc permettra de caractériser facilement tout dérivé carbonylé. Il existe les aldoximes et les cétoximes :
R - CH = O + NH2OH ![]() R - CH = N - OH
R - CH = N - OH
Stéréochimie des oximes :
Les oximes peuvent donner deux stéréoisomères, un E et un Z :
Les oximes peuvent donner deux stéréoisomères, un E et un Z :
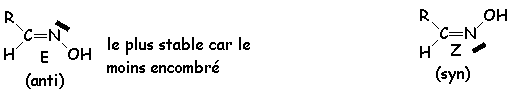
Réaction des oximes :
Les oximes se déshydratent facilement, en présence de SOCl2 ou d'acide sulfurique concentré. Les aldoximes donnent des nitriles :
![]()
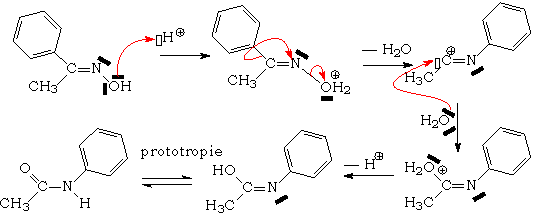
Les cétoximes subissent, en présence des mêmes déshydratants, une réaction de transposition, appelée réarrangement de Beckmann. La plupart du temps, c 'est le substituant situé en anti du groupe OH qui migre :
Équation bilan :
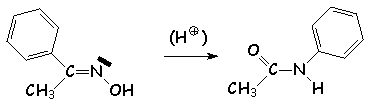
Mécanisme :
La réaction des dérivés carbonylés avec L'hydrazine donne des dérivés cristallisés caractéristiques appelés hydrazones :

Lorsque la cétone est en gros excès, il y a double condensation pour donner une azine :
.gif)
Cas particulier du traitement d'un dérivé carbonylé en milieu fortement basique en présence d'un donneur de protons à très haute température : réaction de WOLFF-KISCHNER

Cette réaction n 'a lieu qu 'avec les dérivés carbonylés (aldéhydes et cétones) et ne se produit pas avec les dérivés d'acide (esters, amides, etc.)
Mécanisme (hors programme) :
.gif)
On constate que L'équilibre est déplacé lors de la formation de L'azote gazeux.
On peut obtenir le même résultat ( réduction d'un dérivé carbonylé en alcane) par la réaction de CLEMMENSEN :
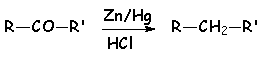
Ces composés donnent de nombreux dérivés cristallisés caractéristiques avec les dérivés carbonylés :
NH2 - NH - CO - NH2 + RCOR ' ® RR 'C = N - NH - CO - NH2
(semicarbazine : quel est l'azote le plus nucléophile) semicarbazone
La 2,4-dinitrophénylhydrazine est le plus connu parmi ces
réactifs et donne des dérivés cristallisés (2,4- dinitrophénylhydrazones) jaunes
avec les dérivés carbonylés saturés et rouges avec les dérivés carbonylés
instaurés :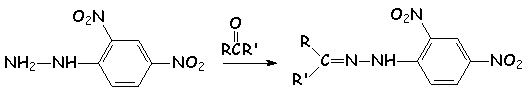
NB : Synthèse de la 2,4-dinitrophénylhydrazine
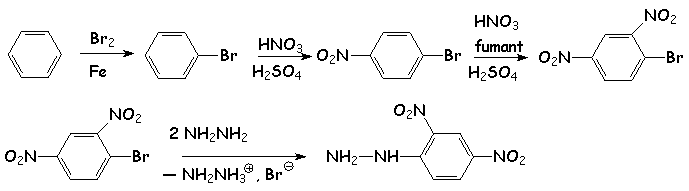
SN aromatique
4. Réactions liées à la mobilité des H en a du carbonyle
4.1. Halogénation des dérivés carbonylés
Il s 'agit de substituer un ou plusieurs H en a du CO par un ou plusieurs halogènes. Cette réaction peut se faire en milieu basique ou acide. Les résultats seront différents.
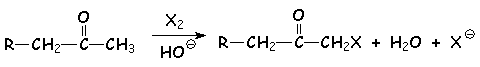
Mécanisme :
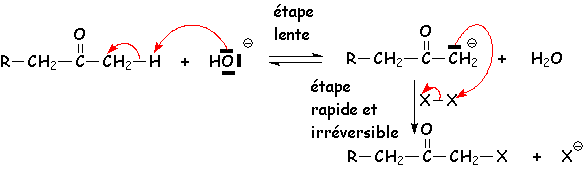
Le proton le plus acide étant celui porté par le carbone le moins substitué par des groupements donneurs, L'halogénation se fera sur la carbone le plus hydrogéné. d'autre part, le premier halogène substitué, par ses effets inductifs attracteurs, va augmenter L'acidité des autres H portés par le même carbone. L'halogénation va donc se poursuivre, en présence d'un excès d'halogène, jusqu 'à remplacement complet de tous les H.
Si la cétone est méthylée, on aboutit à R - CH2 - CO - CX3. Comme nous sommes en milieu basique, ce composé est détruit de la manière qui suit :
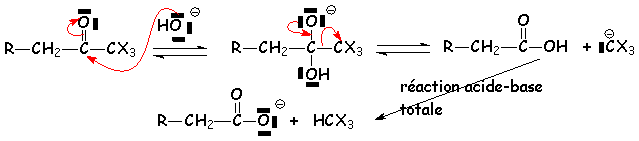
Il s 'agit par exemple de la synthèse officinale du chloroforme :
![]()
L'addition de X2 se fait sur L'énol. C 'est L'énol le plus stable qui se forme donc : le plus substitué par des groupements donneurs (voir la stabilité des alcènes) :
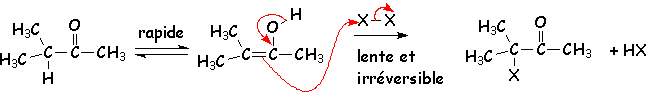
L'halogénation ne se fait qu 'une seule fois en milieu acide.
4.2. Aldolisation - Cétolisation - Crotonisation.
Ici encore, cette réaction peut se faire en milieu basique ou acide. Le produit final n 'est pas toujours identique.
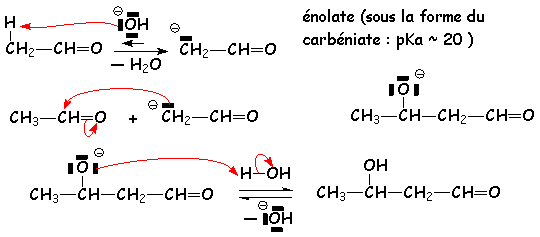
(À température ordinaire)
Le premier équilibre a une constante de 10 -6. Cependant L'équilibre global a une constante supérieure à 1, dans le cas des aldéhydes (6 à 10) ; par contre, dans le cas des cétones, cette constante est de L'ordre de 10 -1. Ceci est dû à la plus faible réactivité du carbonyle des cétones (voir la synthèse des cyanhydrines).
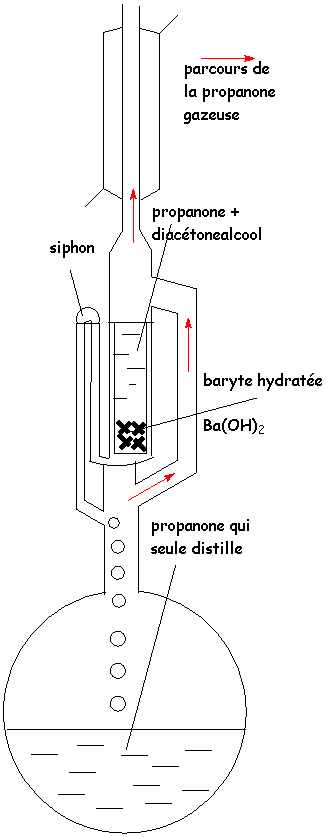
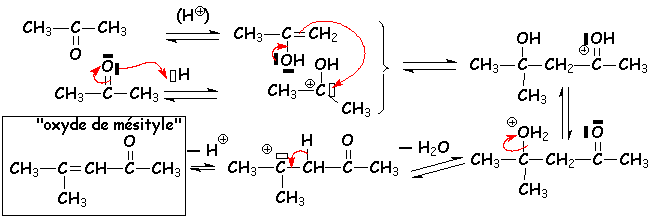
Ainsi, la mise en présence de propanone et de soude, à
température ordinaire, donne à L'équilibre un mélange de 10% de diacétone-alcool
(cétol) 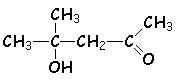 et de 90% de propanone.
et de 90% de propanone.
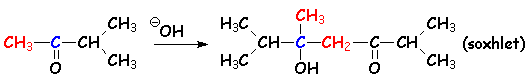
Pour synthétiser avec un bon rendement le diacétone-alcool, on peut utiliser un soxhlet : (ci-contre).
Généralement, à température ordinaire, la réaction s 'arrête à ce stade en milieu basique. Cependant, le chauffage peut favoriser la déshydratation (crotonisation). Celle-ci est difficile, même à chaud, lorsque les structures obtenues sont linéaires.
Par contre, lorsque les doubles liaisons obtenues sont conjuguées avec un cycle aromatique par exemple, la crotonisation a lieu à l'ébullition du solvant, suivant immédiatement L'aldolisation. La réaction est appelée " condensation de Claisen - Schmitt ".
En effet, ces réactions étant toutes équilibrées, il suffit que le produit final soit très stable (ici par conjugaison) pour que la réaction évolue jusqu 'au dérivé carbonylé a,b-éthylénique, appelé aussi a-ènone.
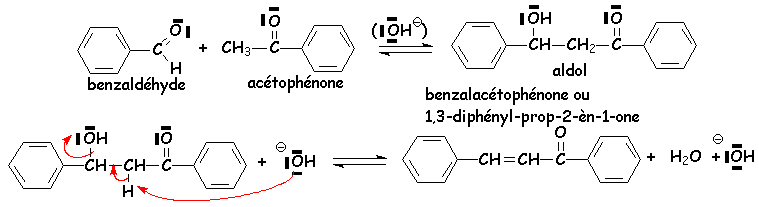
Lorsque la réaction de crotonisation peut être suivie, sans changer de milieu réactionnel, par une réaction conduisant à un produit plus stable, souvent cyclique (non aromatique), elle se fait bien (déplacement d'équilibre). (Voir L'annellation de Robinson).
La crotonisation a toujours lieu en milieu acide, aussi bien avec les aldéhydes et les cétones :
À partir de L'éthanal, on obtient le but-2-ènal ou " crotonaldédyhe ", d'ou le nom de crotonisation : 2 CH3 - CHO ® CH3 - CH = CH - CHO
4.2.3. Régiosélectivité de la réaction d'aldolisation
En milieu basique, c 'est le carbéniate le plus stable (voir 4.1.1.) qui se forme. L'aldolisation se fera en fonction de ce résultat :
En milieu acide, c 'est la stabilité de L'énol qui régit L'orientation de la réaction. Dans le cas précédent (on va jusqu 'à la crotonisation), on obtient :
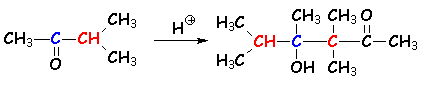
Ici la crotonisation n 'est pas possible, car il ne reste plus d'H en a du carbonyle.
4.2.4. Compétition aldéhyde - cétone.
La cétone et L'aldéhyde fourniront le " carbéniate - énolate ", et L'aldéhyde qui sera sujet de L'attaque nucléophile sur le carbone du carbonyle (voir la synthèse des cyanhydrines pour comprendre la différence de réactivité des aldéhydes et cétones). On obtient donc deux composés majoritaires :
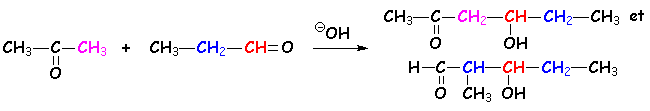
Les a-ènones subiront L'attaque nucléophile des énolates en position 4 (ou b) par rapport au carbonyle :
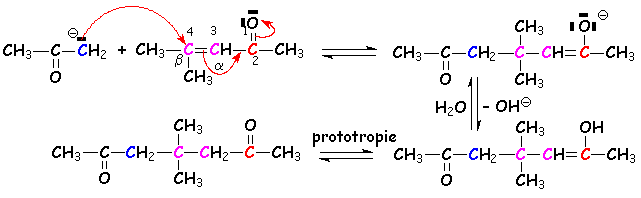
Lorsque, dans le même milieu, le composé dicétonique peut subir une nouvelle aldolisation cyclique, celle-ci se poursuit par une crotonisation, car L'alcène cyclique est stable : c 'est L'annellation de Robinson.
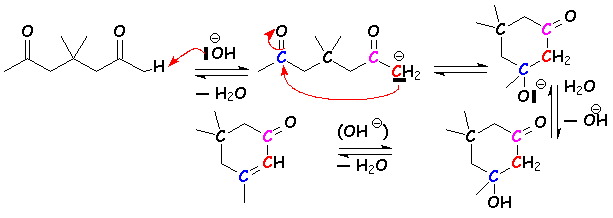
On obtient ici une nouvelle a - ènone.
Rappel : les organométalliques réagissent différemment avec les a - ènones selon la nature du métal :
- les organolithiens donnent une addition 1,2
- les organocuprates R2CuLi donnent une addition 1,4
- les organomagnésiens donnent tantôt L'une, tantôt L'autre, selon L'encombrement des positions 2 et 4
Dans le cas précédent, un organomagnésien donnerait une
addition 1,2, le C4
étant très encombré. Pour obtenir une méthylation en 1,4 , il faut utiliser le
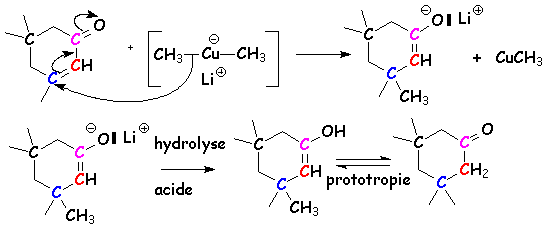
diméthylcuprate de lithium ![]() :
:
Il faut ici une
concentration suffisante en ion énolate / carbéniate, car cet énolate va faire
une SN2 sur un dérivé halogéné. Pour obtenir une mole d'énolate pour
une mole de dérivé carbonylé, il faut utiliser une base plus forte que
lui : L'amidure de sodium (pKa ~ 35) convient. Pour minimiser les
propriétés nucléophiles de cet amidure (qui pourrait substituer L'halogénure
concurremment à L'énolate), on préfère utiliser un amidure substitué tel que le
diisopropylamidure de sodium ou de lithium (LDA) ![]()
Si L'on mélange en proportions stchiométriques, de la cyclohexanone, de L'iodure de méthyle et de L'amidure de sodium, on obtient un mélange de cyclohexanones mono- et polyméthylées en a et a ' :
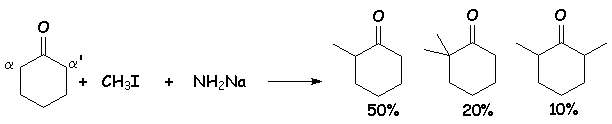
Mécanisme :
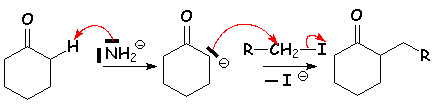
Cette réaction n 'est vraiment intéressante que lorqu 'il s 'agit de " perméthyler " un dérivé carbonylé :
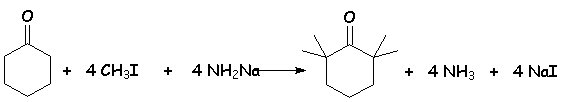
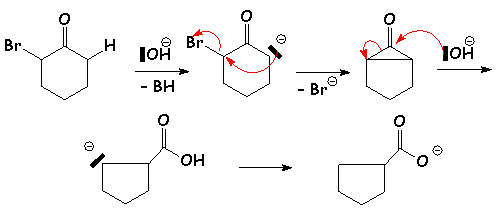
- Transposition de Favorskii (Hors porgramme)
Cette alkylation peut être intramolèculaire, et un cas particulier est celui d'une cétone a-halogénée et possédant un H libre en a' :
Grâce à L'hydrogène porté par CO, les aldéhydes sont bien plus réducteurs que les cétones
Les aldéhydes sont toujours oxydés en acide carboxylique ![]() . De
nombreux oxydants, même faibles, pourront effectuer cette transformation.
. De
nombreux oxydants, même faibles, pourront effectuer cette transformation.
L'oxygène de L'air oxyde lentement les aldéhydes, si bien que les flacons, même bouchés, contiennent une quantité appréciable d'acide carboxylique. La réaction est radicalaire.
L'ion ![]() est un bon
oxydant
est un bon
oxydant ![]() . Il peut donc oxyder les aldéhydes.
. Il peut donc oxyder les aldéhydes.
La demi-équation rédox de cette oxydation est :
![]()
On constate que cette réaction nécessite un milieu basique :
![]()
Or L'ion argent précipite sous forme d'oxyde Ag2O à
ce pH. On stabilisera L'ion argent à pH basique sous la forme d'un complexe
ammoniacal : le diamminoargent ![]() .
.
La demi-équation rédox correspondante est :
![]()
L'équation - bilan est donc :
![]()
L'argent se dépose en film sur les parois du réacteur : c 'est L'expérience du " miroir d'argent ".
L'ion cuivrique ![]() est également un
oxydant faible
est également un
oxydant faible ![]() , qui précipite sous forme d'hydroxyde
, qui précipite sous forme d'hydroxyde ![]() à pH basique. On
le stabilise en le complexant par L'ion tartrate
à pH basique. On
le stabilise en le complexant par L'ion tartrate ![]() sous la forme
d'ion ditartratocuprate +II plan
sous la forme
d'ion ditartratocuprate +II plan
La demi-équation rédox de réduction de ce complexe est la suivante :
![]()
et L'équation bilan
![]()
Cette réaction est importante en biochimie, car elle permet de distinguer les oses (sucres) pouvant exister sous une forme aldéhydique libre (glucose, lactose), et ceux ne le pouvant pas (saccharose).
Ne sont oxydées que par les acides forts oxydants : HNO3 :
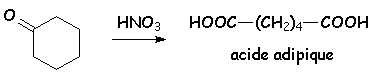
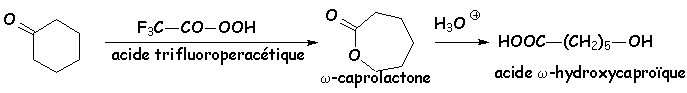
Une réaction industrielle importante utilise les peracides organiques pour transformer les cétones en lactones (esters cycliques) :
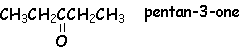
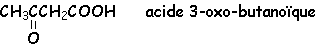
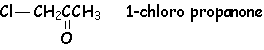
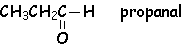
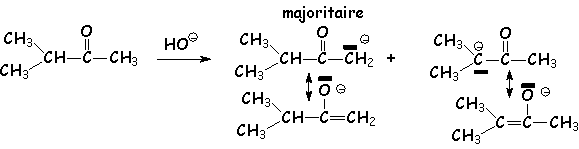
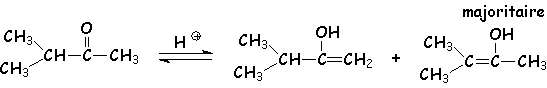
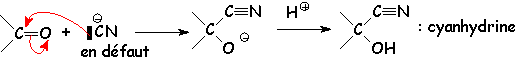
.gif)