Retour à la feuille d'exercices
1. Pas de difficulté particulière. Attention aux molécules présentant un carbone pseudo-asymétrique (1,2,3 et 1,3,5)
2.(CH3)2CBr - CH2Br
3. C6H5 - CH2 - CH2 - CH2Br
4. Se rappeler que pour un même atome, la nucléophilie évolue de la même manière que la basicité.
Pour des atomes différents, cette nucléophilie dépend beaucoup du solvant. Si celui-ci est protique, la liaison H va la masquer d'autant plus que l'atome est électronégatif.
5. Comparer les pouvoirs nucléofuge et nucléophile des divers ions mis en jeu
6. Comparer les pKa de l'acide cyanhydrique et de l'acide chloroéthanoïque
7. Quel est le facteur structural qui défavorise les SN2 ?
8. Les solvolyses sont SN1. Comparer la stabilité des carbocations intermédiaires.
9. Les conditions opératoires font penser à une SN2. Considérer la nature du substrat (encombrement, possibilité d'inversion de Walden, géométrie du carbone fonctionnel).
10. Voir 9.
11. Il est rappelé que la deshydrohalogénation E2 est anticoplanaire
12. Étudier l'influence des substituants du cycle sur la stabilité du carbocation intermédiaire
13. H et Br doivent être en position anticoplanaire
14. idem
15. Un des cycles joue le rôle de nucléophile voisin et substitue l'ion bromure plus rapidement (effet de proximité) que l'ion méthylate, en donnant un carbocation ponté stabilisé par résonance avec le système p du cycle. Puis l'ion méthylate substitue ce gpmnt phényl.
16. On passe par l'intermédiaire :
17. Penser à l'intermédiaire réactionnel ![]()
17bis Comparer la stabilité des "carbocations non classiques" intermédiaires
18. Voir cours
19. La première réaction suit un mécanisme SN2 . Les deux autres un mécanisme SN1. Étudier la stabilité des carbocations pour la seconde question.
20. SN2 et E2. Respecter la stéréochimie liée à ces mécanismes
21. 1. Voir que tous les C fonctionnels sont primaires, et donc que la vitesse diminue avec l'encombrement.
2. Constater que v augmente avec le passage du C fonctionnel de Iaire à IIIaire , ainsi qu'avec l'augmentation de la polarité du solvant.
3. Si v dépend de la nature du nucléophile, la réaction est SN2, etc...
22. SN, d'ordre 1
23. Pourquoi ? : ![]()


3A : E , 3B E (maj) + Z
24. mélange d'éther et d'alcool
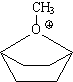
25. résultat à interpréter : 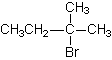
26. Attaque du nucléophile en C4
27 1. C secondaire et nucléophile fort
2. C secondaire et nucléophile fort, solvant peu polaire et protique
3. C secondaire et nucléophile faile, solvant polaire et protique
28 C4H9CN CH2 = CH - CH3 CH3 - C(CH3) = CH - C2H5 hexan-2-ol
29 Utiliser éliminations et additions pour passer des RX aux divers alcènes. Se souvenir que les alcynes peuvent être obtenus par E sur les dérivés dihalogénés.
30 E2 et Würtz
31. 1. SN aromatique 2. Obtention d'un alcène-alcool 3. Réaction acide-base puis SN2 4. Addition d'un carbène
32. L'intermédiaire essentiel est le bromoéthane. On utilise ensuite des SN, des synthèses organométalliques (magnésiens, lithiens, cadmiens, würtz,...)
33 Où doit être positionnée la fonction alcool de X pour que la déshydratation ne donne qu'un seul composé ?
34. Addition d'HBr puis E2
35. L'un des composés est chiral, l'autre est achiral
36. Le pouvoir rotatoire d'un mélange est égal à la somme des pouvoirs rotatoires. On fait apparaître les fractions molaires en utilisant les pouvoirs rotatoires spécifiques : on trouve x1S = 5/6, en considérant que le mélange est formé de racémique et de S . Pour la question 3, voir que le racémique donne toujours du racémique, qu'il y ait inversion ou racémisation.
37. Où se fait l'attaque du nucléophile lors de l'inversion du pouvoir rotatoire spécifique?
38. Quelle est la réaction la plus rapide dans ce cas-là ? Quel va être alors le nucléophile ?