1.1. Étude de l'élément.
1.1.1. Configuration électronique
![]() . C’est un métal de
transition. Les orbitales d sont environ à moitié remplies. La bande
d sera donc à moitié remplie, presque tous les électrons remplissant les
O.M. liantes : la liaison métallique sera donc très forte, ce qui entraîne
également une forte compacité (et donc une masse volumique élevée : 7900
kg.m–3.
. C’est un métal de
transition. Les orbitales d sont environ à moitié remplies. La bande
d sera donc à moitié remplie, presque tous les électrons remplissant les
O.M. liantes : la liaison métallique sera donc très forte, ce qui entraîne
également une forte compacité (et donc une masse volumique élevée : 7900
kg.m–3.
Le rayon métallique vaut 0,126 nm ; le rayon ionique de Fe2+ vaut 0,076 nm, celui de Fe3+ vaut 0,067 nm.
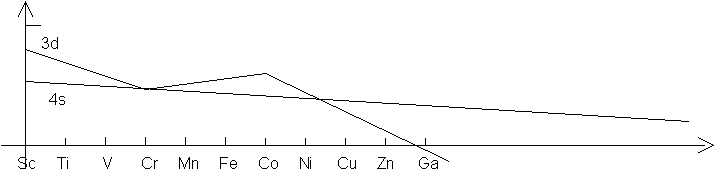
Le diagramme suivant montre l’évolution de l’énergie des électrons 3d et 4s lorsqu’on passe du scandium (Sc) au gallium (Ga) :
On voit que cette énergie devient de
plus en plus négative, et qu’il sera plus difficile d’ioniser les métaux de
droite que de gauche. Ainsi ![]() et
et ![]() sont-ils pratiquement ioniques et contiennent des ions
sont-ils pratiquement ioniques et contiennent des ions
![]() et
et ![]() . Par contre
l’ion ferrate
. Par contre
l’ion ferrate ![]() (
(![]() ) est purement covalent, et
) est purement covalent, et ![]() n’existe pas. Dans les
périodes inférieures, la taille des ions métalliques aidant, les composés du
type
n’existe pas. Dans les
périodes inférieures, la taille des ions métalliques aidant, les composés du
type ![]() existent (
existent (![]() et
et ![]() ).
).
Les électrons du fer sont
relativement faciles à arracher: les deux électrons 4s et un seul 3d (pour
obtenir la configuration 3d5, la plus stable car les OA d sont
exactement à moitié remplies): ![]() . L’électron 3d est plus difficile à arracher, à
cause de la petitesse de l’ion :
. L’électron 3d est plus difficile à arracher, à
cause de la petitesse de l’ion : ![]() .
.
Plus généralement, voici les
potentiels rédox pour les couples ![]() :
:
|
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu | |
|
|
–1,6 |
–1,18 |
–0,91 |
–1,18 |
–0,44 |
–0,28 |
–0,24 |
+0,34 |
|
|
–0,37 |
–0,25 |
–0,41 |
+1,54 |
+0,77 |
+1,84 |
/ |
/ |
On y retrouve la stabilité
particulière des états d5 pour ![]() et
et ![]() .
.
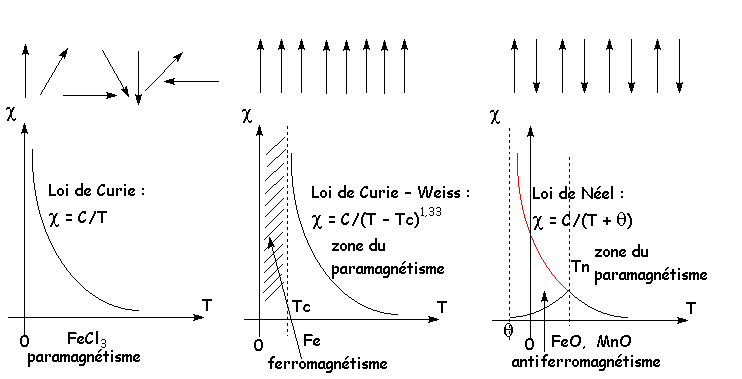
La plupart des composés d’éléments de transition sont paramagnétiques, et leurs propriétés ont souvent été déduites à partir de ces données. Étudions ce magnétisme en détail.
La susceptibilité magnétique d’une substance présentant un moment cinétique électronique J = L + S (L = moment cinétique orbital et S = moment cinétique de spin), est égale à :
![]()
où ![]() est le nombre
d’Avogadro ; g, le facteur de Landé de l’atome (2 en général) ;
est le nombre
d’Avogadro ; g, le facteur de Landé de l’atome (2 en général) ; ![]() , le magnéton de Bohr
=
, le magnéton de Bohr
= ![]() ; k
est la constante de Boltzmann =
; k
est la constante de Boltzmann = ![]() . A température ordinaire, kT "
. A température ordinaire, kT " ![]() , donc th (x) = x ,
soit
, donc th (x) = x ,
soit ![]() .
Pour les atomes de la première période de transition, J = S , les valeurs
propres de S étant égales à
.
Pour les atomes de la première période de transition, J = S , les valeurs
propres de S étant égales à ![]() . Par exemple
. Par exemple ![]() possède une somme de
spin égale à 2,5. À 300K, la valeur calculée de c vaut 0,438
possède une somme de
spin égale à 2,5. À 300K, la valeur calculée de c vaut 0,438 ![]() , la valeur
expérimentale valant 0.430
, la valeur
expérimentale valant 0.430 ![]() .
.
Le diamagnétisme est une propriété
de toutes les formes de matière. Car toutes les substances possèdent des
orbitales remplies pour lesquelles ![]() = 0 et donc
l = 0. Donc pas de paramagnétisme s’il n’existe pas d’électrons
célibataires. Cependant, dans un champ magnétique B , un petit moment
magnétique, proportionnel et opposé à B , apparaît. Le paramagnétisme est 100
fois plus fort que le diamagnétisme. Celui-ci ne se voit donc pas pour une
substance paramagnétique.
= 0 et donc
l = 0. Donc pas de paramagnétisme s’il n’existe pas d’électrons
célibataires. Cependant, dans un champ magnétique B , un petit moment
magnétique, proportionnel et opposé à B , apparaît. Le paramagnétisme est 100
fois plus fort que le diamagnétisme. Celui-ci ne se voit donc pas pour une
substance paramagnétique.
De quoi s’agit-il? On peut montrer
que sous l’effet de B , il apparaît un moment magnétique ![]() où
où ![]() est la valeur moyenne
du rayon de l’orbite de l’électron considéré. Par exemple, pour un électron 1s,
est la valeur moyenne
du rayon de l’orbite de l’électron considéré. Par exemple, pour un électron 1s,
![]() étant
le rayon de Bohr.
étant
le rayon de Bohr.
Nous avons vu qu’une substance paramagnétique voyait sa susceptibilité magnétique varier proportionnellement à 1/T (loi de Curie). Pour certaines substances, on constate qu’au dessous d’une certaine température, le comportement paramagnétique disparaît et fait place à une zone où existe une aimantation spontanée, qui augmente très fortement dans un champ magnétique extérieur. Cette température est la température de Curie et la susceptibilité paramagnétique suit alors la loi de CurieWeiss :
![]()
La forte augmentation de l'
aimantation au dessous de ![]() provient de l’alignement les uns par rapport aux
autres des moments magnétiques des atomes. Au-dessous de
provient de l’alignement les uns par rapport aux
autres des moments magnétiques des atomes. Au-dessous de ![]() , l’agitation thermique
ne peut vaincre la force magnétique qui maintient l’alignement des moments
magnétiques des atomes. Il y a ferromagnétisme. Pour certaines substances, les
forces entre les petits moments sont telles qu’il vont s’aligner deux à deux de
manière antiparallèle. On comprend que cet agencement, existant au dessous d’une
température dite de Néel, va au contraire diminuer la susceptibilité magnétique
jusqu’à l’annuler presque au zéro absolu.
, l’agitation thermique
ne peut vaincre la force magnétique qui maintient l’alignement des moments
magnétiques des atomes. Il y a ferromagnétisme. Pour certaines substances, les
forces entre les petits moments sont telles qu’il vont s’aligner deux à deux de
manière antiparallèle. On comprend que cet agencement, existant au dessous d’une
température dite de Néel, va au contraire diminuer la susceptibilité magnétique
jusqu’à l’annuler presque au zéro absolu.
La magnétite ![]() est l’exemple typique
de composé ferrimagnétique. On peut rappeler que sa structure est celle d’une
maille CFC d’ions
est l’exemple typique
de composé ferrimagnétique. On peut rappeler que sa structure est celle d’une
maille CFC d’ions ![]() , les ions
, les ions ![]() étant situés dans le ¼ des sites octaédriques, et les
ions
étant situés dans le ¼ des sites octaédriques, et les
ions ![]() pour moitié dans ¼ des sites octaèdriques, et pour moitié dans
pour moitié dans ¼ des sites octaèdriques, et pour moitié dans ![]() des sites
tétraèdriques. Il s’agit d’un spinelle inverse. Il y a alignement des moments
magnétiques dans chaque type de site : (-) dans les tétraèdriques et (+) dans
les octaèdriques. On a donc la disposition suivante pour les petits
moments:
des sites
tétraèdriques. Il s’agit d’un spinelle inverse. Il y a alignement des moments
magnétiques dans chaque type de site : (-) dans les tétraèdriques et (+) dans
les octaèdriques. On a donc la disposition suivante pour les petits
moments:
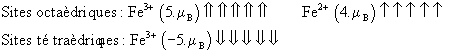
On constate que les moments
magnétiques des ions ferriques s’annulent. Ne restent que les moments
magnétiques des ions ferreux, qui confèrent à la magnétite une aimantation
permanente proportionnelle au seul état de spin des ![]() .
.
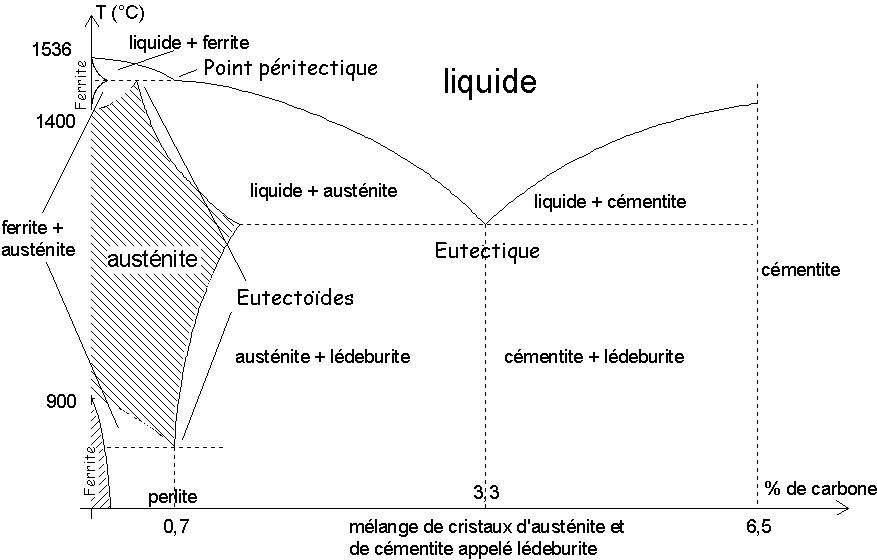
1.3.1. Structure.
Le fer existe sous trois formes:
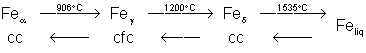
Il représente 4,7% de la croûte
terrestre. Il est majoritaire dans le noyau. Les principaux minerais sont
l’hématite ![]() , la magnétite
, la magnétite ![]() , la limonite
, la limonite ![]() , la sidérite
, la sidérite ![]() ...
...
-solutions solides de substitution :
Cr, la structure est la même (a , g , d ). On l’utilise pour obtenir l’acier chromé, l’inox...
Ni, c’est la même chose.,
-solutions solides d’insertion :
surtout avec le carbone. Ainsi le fer a donne la ferrite, le fer g l’austénite.
![]() est la
cémentite.
est la
cémentite.
Le fer est ferromagnétique, mou pour le fer a et d , dur pour le fer g Il est tenace et ductile.
Il est relativement stable à l' air.
S’oxyde à l’air humide (voir le § corrosion). Il réagit avec les oxydants à
chaud (oxygène, chlore) et réduit les oxydants en solution aqueuse (![]() )
)
2. Étude des propriétés des ions ferreux et ferriques.
2.1. Les complexes du fer.
2.1.1. Structure.
Voici ci-dessous un tableau donnant les différents types de complexes du fer.
|
|
remplissage |
structure |
exemples |
|
– II |
|
tétraèdrique |
|
|
0 |
|
trigonal bipyramidal |
|
|
+ I |
|
octaèdrique |
|
|
+ II |
|
tétraèdrique octaèdrique |
|
|
+ III |
|
tétraèdrique trigonal bipyramidal octaèdrique |
|
|
+ IV |
|
octaèdrique |
|
|
+ VI |
|
tétraèdrique |
|
Nous nous intéressons aux états II et III. La structure électronique de ces complexes dépend de la nature du ligand. Si le champ cristallin est grand, le complexe est à spin faible; s’il est petit, le complexe est à spin fort et toujours paramagnétique.
Nous devons rappeler la série spectrochimique :
![]()
La couleur des complexes dépend de
la force du champ. Ainsi ![]() est-il orange, car il absorbe dans le bleu-vert. Si l’hydroxyde
ferreux (qui devait être blanc) apparaît vert, c’est à cause des traces
d’hydroxyde ferrique qui réémettent le bleu-vert. Par contre ,
est-il orange, car il absorbe dans le bleu-vert. Si l’hydroxyde
ferreux (qui devait être blanc) apparaît vert, c’est à cause des traces
d’hydroxyde ferrique qui réémettent le bleu-vert. Par contre , ![]() est incolore (absorbe
dans l’UV). Parfois l’excitation lumineuse provoque un transfert de charges du
ligand au métal, par passage d’électrons d’une orbitale p pleine du ligand à une
orbitale d de bonne symétrie du métal. Quelques exemples :
est incolore (absorbe
dans l’UV). Parfois l’excitation lumineuse provoque un transfert de charges du
ligand au métal, par passage d’électrons d’une orbitale p pleine du ligand à une
orbitale d de bonne symétrie du métal. Quelques exemples :
![]() bleu, par transfert
de charge.
bleu, par transfert
de charge.
![]() ,rouge sang
,rouge sang
Le fer II et le fer III forment des complexes qui sont pour la plupart octaèdriques. Il est intéressant d’étudier la stabilité relative des complexes II et III (potentiels rédox), ainsi que les réactions d’élimination et de substitution qui ont lieu sur ces complexes.
Le potentiel redox du couple ![]() vaut 0,77V.
La substitution des ligands
vaut 0,77V.
La substitution des ligands ![]() par d’autres ligands, en modifiant la stabilité
relative des complexes, modifie le potentiel redox. Par exemple pour le couple
par d’autres ligands, en modifiant la stabilité
relative des complexes, modifie le potentiel redox. Par exemple pour le couple
![]() ,
, ![]() . Cela montre
que l’hexacyanoferrate+III n’est qu’un petit peu moins stable que
l’hexacyanoferrate +II , alors que l’ion hexaaquofer+III est
nettement moins stable que l’ion hexaaquofer+II . Autre exemple,
celui du couple triorthophénanthrolinofer+III /
triorthophénanthrolinofer+II , dont le potentiel redox vaut 1,12
V.
. Cela montre
que l’hexacyanoferrate+III n’est qu’un petit peu moins stable que
l’hexacyanoferrate +II , alors que l’ion hexaaquofer+III est
nettement moins stable que l’ion hexaaquofer+II . Autre exemple,
celui du couple triorthophénanthrolinofer+III /
triorthophénanthrolinofer+II , dont le potentiel redox vaut 1,12
V.
orthophénanthroline : 
Lorsqu’un ligand peut céder un de ses atomes ou ions en présence d’un accepteur (proton en présence d’une base par exemple), on est en présence d’une réaction d’élimination, qui, dans l’exemple cité, est une réaction acide-base :
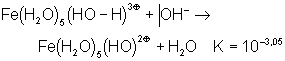
Pour le couple
![]()
Ce sont les plus importantes. La
substitution peut être d’ordre 1 (avec départ préalable d’un ligand) ou d’ordre
2 (avec addition élimination, comme sur les carbonylés en orga). La stabilité
des complexes est donnée par leur constante de dissociation Kd, relative à la
substitution d’un ou plusieurs ligands par ![]() . La vitesse des réactions dépend de la capacité d’un
ligand à être expulsé du complexe : c’est la labilité du ligand. Par exemple,
. La vitesse des réactions dépend de la capacité d’un
ligand à être expulsé du complexe : c’est la labilité du ligand. Par exemple,
![]() sont
peu labiles, alors qu’
sont
peu labiles, alors qu’![]() est très labile. Un complexe peut avoir un Kd supérieur à 1 et
exister dans l’eau à cause de la faible labilité du ligand. On peut ainsi
étudier en solution aqueuse pendant un temps assez long le complexe
est très labile. Un complexe peut avoir un Kd supérieur à 1 et
exister dans l’eau à cause de la faible labilité du ligand. On peut ainsi
étudier en solution aqueuse pendant un temps assez long le complexe ![]() avant qu’il
ne soit détruit.
avant qu’il
ne soit détruit.
Les complexes les plus stables sont
les complexes cyanés. Cependant, la labilité de ![]() est plus importante
dans l’hexacyanoferrate+III que dans l’hexacyanoferrate+II
. Aussi la solution aqueuse du premier est-elle plus toxique, car elle contient
un peu d’ions cyanure remplacés par l’eau dans le complexe.
est plus importante
dans l’hexacyanoferrate+III que dans l’hexacyanoferrate+II
. Aussi la solution aqueuse du premier est-elle plus toxique, car elle contient
un peu d’ions cyanure remplacés par l’eau dans le complexe.
![]() réagit sur
réagit sur ![]() pour donner
le bleu de Turnbull,
pour donner
le bleu de Turnbull, ![]() réagit sur
réagit sur ![]() pour donner le bleu de Prusse.
pour donner le bleu de Prusse.
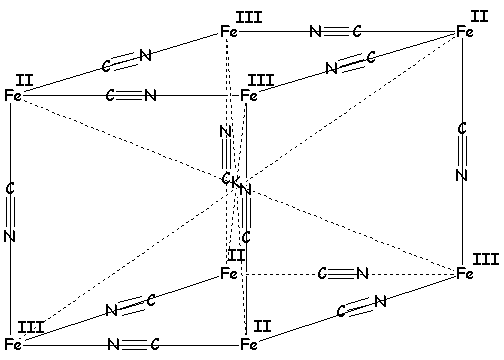
Ces deux pigments sont en réalité
identiques et ont pour formule brute : ![]() , dont voici la
structure cristalline :
, dont voici la
structure cristalline :
Fe+III donne beaucoup de complexes stables avec des composés oxygénés, par exemple le triacétylacétonatofer+III , qui a une couleur rouge intense caractéristique du fer :
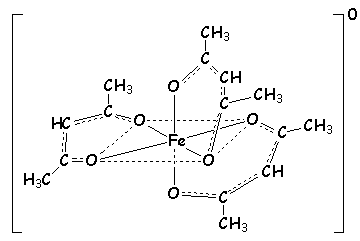
Les ions thiocyanate SCN– donnent une série de complexes rouges où les ions SCN– sont très labiles, puisque l’ajoût de F– fait disparaître instantanément la couleur par formation de complexes fluorés très stables.
A l’état solide, on trouve beaucoup
de complexes hexacoordinés, par exemple ![]() , et surtout les
seuls complexes tétraèdriques :
, et surtout les
seuls complexes tétraèdriques : ![]() , ou encore
, ou encore ![]() .
.
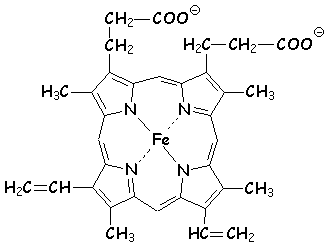
Le plus important est l’hème, où Fe +II est complexé par des azote. L’action de NO ou des nitrites oxyde irréversiblement Fe +II en Fe +III . C’est la méthémoglobine, incapable d’opérer le transport de l’oxygène et du gaz carbonique dans le sang. Fe+II demeure dans cet état d’oxydation lors du chargement d’oxygène dans les poumons, il y est simplement complexé par O2 .
Leur particularité réside dans leur
facilité à être hydrolysés en solution aqueuse. Cela est dû au caractère
polarisant important des cations (rayons ioniques faibles). Les ions ![]() et
et ![]() ont des
liaisons métal-oxygène si fortes qu’en présence de base, même faible, les
liaisons O–H ont un caractère acide important.
ont des
liaisons métal-oxygène si fortes qu’en présence de base, même faible, les
liaisons O–H ont un caractère acide important.
Ce sont aussi les propriétés
réductrices de la solution de ![]() qui retiennent l’attention. C’est pourquoi, exposée à
l’air, cette solution est colorée par des traces de
qui retiennent l’attention. C’est pourquoi, exposée à
l’air, cette solution est colorée par des traces de ![]() .
.
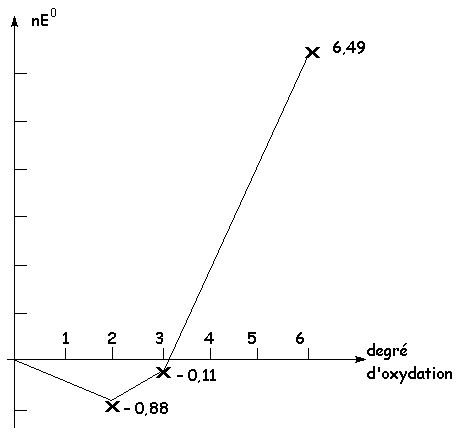
Données : ![]()
![]()
![]()
![]()
2.3.1. Aspect thermodynamique.
Le système ![]() est un système rapide,
par contre les systèmes oxydants sont lents : surtension de
est un système rapide,
par contre les systèmes oxydants sont lents : surtension de ![]() sur Fe = 1 V, celle de
sur Fe = 1 V, celle de
![]() sur Fe
= 0,1 V. De la concentration de la solution dépend sa résistance, qui fait
apparaître une chute de tension. Si la résistance est infinie (par exemple dans
le cas d’un métal couvert d’oxyde), l’intensité de corrosion devient
nulle.
sur Fe
= 0,1 V. De la concentration de la solution dépend sa résistance, qui fait
apparaître une chute de tension. Si la résistance est infinie (par exemple dans
le cas d’un métal couvert d’oxyde), l’intensité de corrosion devient
nulle.
Les systèmes qui nous intéressent
sont donc : ![]() ,
, ![]() et
et ![]() . Sur le fer, la surtension de l’oxygène vaut environ 1,4 V ;
celle de l’hydrogène vaut 0,1 V. Le premier couple est un système rapide . Voici
le diagramme I/E à pH = 7 :
. Sur le fer, la surtension de l’oxygène vaut environ 1,4 V ;
celle de l’hydrogène vaut 0,1 V. Le premier couple est un système rapide . Voici
le diagramme I/E à pH = 7 :
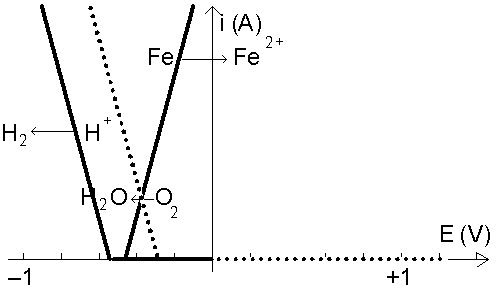
On constate bien qu’à pH = 7, c’est l’oxygène qui oxyde le fer. Certes l’intensité de corrosion, donc la vitesse de la réaction est faible, mais à long terme, le fer sera malgré tout corrodé. Il faut donc diminuer cette intensité. Nous disposons pour cela de deux possibilités : soit diminuer le potentiel du milieu, soit diminuer la valeur absolue de la pente de la droite I = f(E), en augmentant la résistance du milieu.
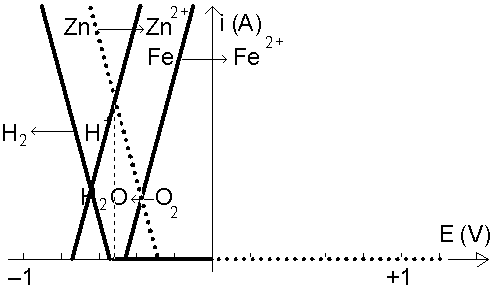
C’est l’oxygène qui oxyde le zinc, et cette réaction impose le potentiel, qui est inférieur à –0,44 V ; donc, le fer ne peut plus être oxydé à ce potentiel et est protègé (protection des coques de navires).
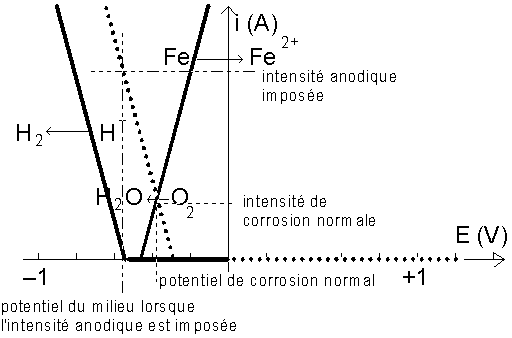
Lorsque l’on place un générateur de courant aux bornes de nos électrodes, on constate que le potentiel du milieu descend au dessous de –0,44 V, ce qui protège le fer.
Ceux-ci se fixent sur la cathode et augmentent fortement la résistance de l’électrolyte, d’où diminution de l’intensité de corrosion :
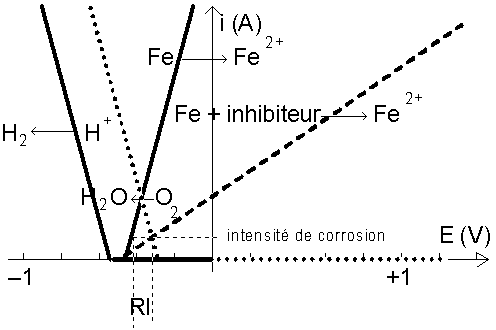
La peinture d’une plaque métallique fait tendre la résistance de l’électrolyte vers l’infini, ce qui fait tendre l’intensité de corrosion vers zéro.
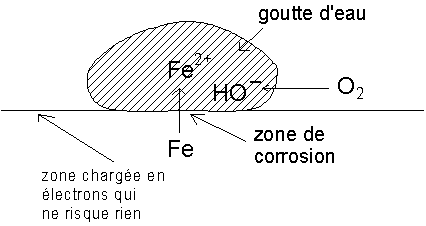
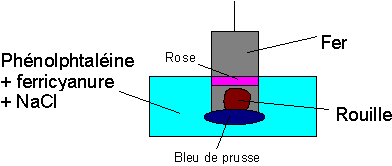
D’après le schéma de la goutte d’eau, on constate que la corrosion se fait là où il n’y a pas d’oxygène, à la cathode. C’est ainsi que la rouille, lorsqu’elle apparaît en un point d’une carrosserie de voiture, a déjà beaucoup progressé sous la peinture adjacente.
3.1. Structure. Non stœchiométrie.
A température ordinaire, les seuls
oxydes de fer stables sont l’hématite ![]() et la magnétite
et la magnétite ![]() . Leurs
structures sont respectivement celles du corindon (alumine a ) et d’une spinelle
inverse. La réduction de de la magnétite au dessus de 1171K fait d’abord
apparaître un oxyde de fer . La réduction se poursuit sans qu’il y ait ni de
modification de phase, ni de modification de maille, jusqu’à ce que cet oxyde
ait la composition Fe24O25. Puis la réduction fait
apparaître du fer pur, sans qu’il y ait jamais apparition de FeO
stœchiométrique.
. Leurs
structures sont respectivement celles du corindon (alumine a ) et d’une spinelle
inverse. La réduction de de la magnétite au dessus de 1171K fait d’abord
apparaître un oxyde de fer . La réduction se poursuit sans qu’il y ait ni de
modification de phase, ni de modification de maille, jusqu’à ce que cet oxyde
ait la composition Fe24O25. Puis la réduction fait
apparaître du fer pur, sans qu’il y ait jamais apparition de FeO
stœchiométrique.
Comment expliquer ce phénomène ? On a d’abord constaté que l’oxyde ferreux était un semi-conducteur de type "p". Pour obtenir la formule Fe24O25 à partir de FeO, il faut faire la réaction théorique suivante :
![]() Ž
Ž![]()
Le signe Ž représente un défaut lacunaire dans le réseau cationique. Ces lacunes représentent des trous positifs dans la bande de valence de l’oxyde ferreux, d’où ses propriétés de semi-conducteur.de type "p".
Si la pression d’oxygène augmente encore, de plus en plus d’ions ferreux sont oxydés en ions ferriques, avec augmentation du nombre de lacunes, jusqu’à l’obtention de Fe21O25:
![]() Ž
Ž![]()
La création de ces lacunes va modifier la couleur de FeO. Celui-ci devrait être blanc. Il est noir, car ces lacunes, appelées centre F, vont absorber dans tout le spectre du visible. De même, NiO stœchiomètrique est vert, alors que NiO non stœchiomètrique est noir.
Une étude thermodynamique permet de montrer que le composé non stœchiométrique est plus stable que FeO théorique, car la création de lacunes ordonnées est parfois la source d’une diminution d’énergie libre, par augmentation d’entropie.
Voir diagramme d’Ellingham.