Spectroscopie infrarouge
Les transitions énergétiques se font ici entre les niveaux d’énergie de rotation des molécules ou entre leurs niveaux d’énergie de vibration.
Les transitions entre niveaux de rotation
apparaissent dans l’I.R. lointain (de 20 à 250 mm ou de 500 à 40 ![]() ). Les transitions entre niveaux vibrationnels
apparaissent de 1 à 20 mm (ou de 10 000 à 500
). Les transitions entre niveaux vibrationnels
apparaissent de 1 à 20 mm (ou de 10 000 à 500 ![]() ). Nous ferons porter notre étude sur les transitions
vibrationnelles. On constate qu’elles nécessitent plus d’énergie que les
transitions rotationnelles. Aussi la lumière excitatrice provoquera-t-elle, pour
chaque transition vibrationnelle, une multitude de transitions rotationnelles,
qui vont donner au pic de transition vibrationnelle l’allure d’une bande
d’absorption :
). Nous ferons porter notre étude sur les transitions
vibrationnelles. On constate qu’elles nécessitent plus d’énergie que les
transitions rotationnelles. Aussi la lumière excitatrice provoquera-t-elle, pour
chaque transition vibrationnelle, une multitude de transitions rotationnelles,
qui vont donner au pic de transition vibrationnelle l’allure d’une bande
d’absorption :
1. Élongation.
Appelé aussi vibration de valence ou "stretching", ce mode concerne la vibration de la molécule le long des liaisons. La fréquence de l’onde électromagnétique qui induit la vibration d’élongation est donnée par la relation :
![]()
où k est la constante de force de la liaison
(considérée ici comme un ressort), proportionnelle à l’énergie de liaison, et m
la masse réduite des deux atomes reliés par cette liaison. Ainsi, les liaisons
multiples, plus énergétiques que les simples, auront une constante de force plus
élevée, donc une fréquence de vibration (remplacée dans la pratique par le
nombre d’onde) plus élevée que celles des liaisons simples entre atomes
identiques : C–C absorbe vers 1100 ![]() , C=C
vers 1600
, C=C
vers 1600 ![]() et
Cº C vers 2100
et
Cº C vers 2100 ![]() . Par contre, les liaisons X–H, où X est un
atome quelconque (C, N, O, ...), auront une fréquence d’élongation plus élevée
que celle d’une liaison C–X, car la masse réduite m y est plus petite : pour
C–H, m = 0,92 u.a. ; pour C–C,
m = 6 u.a.
. Par contre, les liaisons X–H, où X est un
atome quelconque (C, N, O, ...), auront une fréquence d’élongation plus élevée
que celle d’une liaison C–X, car la masse réduite m y est plus petite : pour
C–H, m = 0,92 u.a. ; pour C–C,
m = 6 u.a.
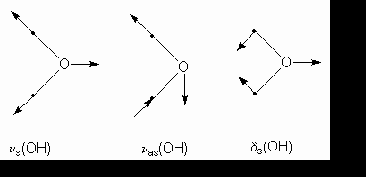
2. Déformations dans et hors du plan.
Considérons une structure ![]() . En plus de la vibration de
valence, l’angle des liaisons peut varier : il y a flexion ou déformation. Ces
déformations peuvent avoir lieu dans le plan des deux liaisons concernées (on
les note d )
ou hors du plan (on les note g ou r ). Il y a aussi possibilité de
déformations symétriques et asymétriques. voici quelques exemples :
. En plus de la vibration de
valence, l’angle des liaisons peut varier : il y a flexion ou déformation. Ces
déformations peuvent avoir lieu dans le plan des deux liaisons concernées (on
les note d )
ou hors du plan (on les note g ou r ). Il y a aussi possibilité de
déformations symétriques et asymétriques. voici quelques exemples :
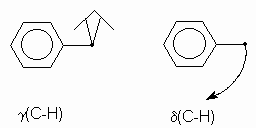
Application de l’I.R. à la détermination des diverses fonctions d’un composé organique.
Non seulement la nature des deux atomes vibrants intervient dans la valeur de la constante de force, mais aussi l’environnement électronique. Aussi chaque groupement fonctionnel aura-t-il des fréquences caractéristiques d’élongation et de déformation. Nous allons passer en revue les diverses fonctions grâce à l’étude de quelques spectres :
1. Les groupements carbonés saturés : les alcanes.
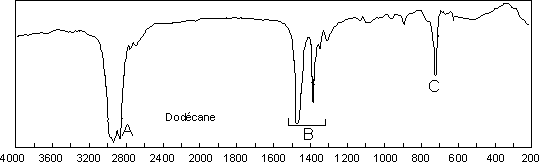
On trouve principalement les vibrations
d’élongation de la liaison C–H entre 3000 et 2840 ![]() . Nous
retrouvons ici les fréquences suivantes :
. Nous
retrouvons ici les fréquences suivantes :
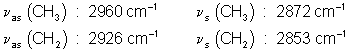
Il suffira de repérer une absorption dans ce domaine pour suspecter fortement la présence de liaisons C–H.
Vers 1400 ![]() se situent les
vibrations de déformation dans le plan des liaisons C–H :
se situent les
vibrations de déformation dans le plan des liaisons C–H :
![]()
Une vibration de déformation hors du plan des ![]() apparaît à 722
apparaît à 722 ![]() . Les
. Les ![]() sont
très faibles et se situent entre 1200 et 1800
sont
très faibles et se situent entre 1200 et 1800 ![]() .
.
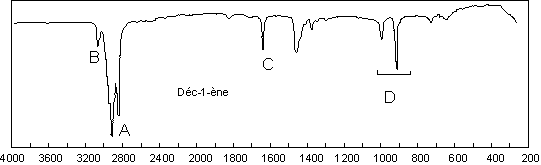
2. Doubles liaisons carbone - carbone.
Par rapport à l’exemple précédent, il apparaît
deux pics nouveaux : à 1645 ![]() , il s’agit de
, il s’agit de
![]() . À 3050
. À 3050 ![]() , il s’agit de
, il s’agit de
![]() . Les vibrations des groupements
saturés apparaissent toujours, et il faut encore remarquer les deux bandes
. Les vibrations des groupements
saturés apparaissent toujours, et il faut encore remarquer les deux bandes ![]() à 986 et 907
à 986 et 907 ![]() . Ces deux
bandes ne sont à étudier que s’il y a un problème de stéréochimie éthylénique (Z
ou E) non soluble par ailleurs.
. Ces deux
bandes ne sont à étudier que s’il y a un problème de stéréochimie éthylénique (Z
ou E) non soluble par ailleurs.
Lorsque les doubles liaisons sont conjuguées :
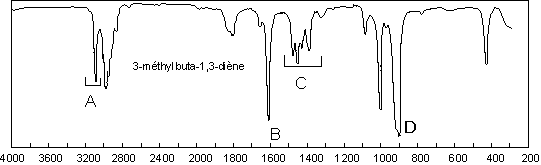
Les trois bandes précédentes subissent un effet
hyperchrome ; le ![]() subit
en outre un effet hypsochrome et les autres
subit
en outre un effet hypsochrome et les autres ![]() un effet bathochrome :
un effet bathochrome :
3. Triple liaison carbone–carbone.
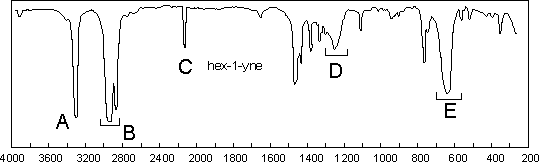
Il faut remarquer la faible bande de l’élongation
![]() à 2110
à 2110 ![]() . On ne la voit
pas toujours, surtout lorsqu’il s’agit d’alcynes disubstitués. Par contre, la
bande d’élongation
. On ne la voit
pas toujours, surtout lorsqu’il s’agit d’alcynes disubstitués. Par contre, la
bande d’élongation ![]() des
alcynes monosubstitués est toujours intense et sort ici à 3268
des
alcynes monosubstitués est toujours intense et sort ici à 3268 ![]() . Moins
importante à signaler est la bande de déformation de
. Moins
importante à signaler est la bande de déformation de ![]() acétylénique
(630
acétylénique
(630 ![]() ) , ainsi que son premier harmonique (1247
) , ainsi que son premier harmonique (1247 ![]() ).
).
4. Composés aromatiques mononucléaires (benzènoïdes).
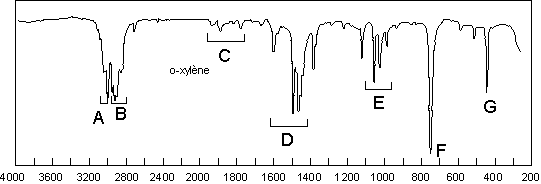
Il faut toujours s’intéresser aux bandes des
basses fréquences : de 900 à 650 ![]() . C’est là que
l’on trouve les renseignements concernant le nombre de substituants du cycle
aromatique et leur position l’un par rapport à l’autre. Sur notre exemple,
l’unique bande de déformation hors du plan de la liaison
. C’est là que
l’on trouve les renseignements concernant le nombre de substituants du cycle
aromatique et leur position l’un par rapport à l’autre. Sur notre exemple,
l’unique bande de déformation hors du plan de la liaison ![]() aromatique
aromatique
![]() montre l’existence d’une disubstitution –1,2 ;
et ce d’autant plus sûrement qu’il s’agit d’une bande intense.
montre l’existence d’une disubstitution –1,2 ;
et ce d’autant plus sûrement qu’il s’agit d’une bande intense.
Dans l’exemple suivant (alcool benzylique avec
cycle monosubstitué), on trouve deux bandes fortes ![]() , correspondant aux deux modes privilégiés de déformation
hors du plan pour 5 hydrogènes aromatiques adjacents. On trouve, dans la zone
allant de 1300 à 1000
, correspondant aux deux modes privilégiés de déformation
hors du plan pour 5 hydrogènes aromatiques adjacents. On trouve, dans la zone
allant de 1300 à 1000 ![]() les bandes de déformation dans le plan des H
aromatiques. Elles sont plutôt faibles et nous ne nous en serviront pas pour la
détermination fonctionnelle.
les bandes de déformation dans le plan des H
aromatiques. Elles sont plutôt faibles et nous ne nous en serviront pas pour la
détermination fonctionnelle.
Intéressante aussi est la zone comprise entre 2000
et 1667 ![]() (lorsqu’il n’y a pas de carbonyle dans la molécule) où l’on
retrouve les harmoniques des bandes de déformation hors du plan et dans le plan
: c’est la signature de la molécule aromatique, qui peut confirmer, si
nécessaire, les informations obtenues grâce aux
(lorsqu’il n’y a pas de carbonyle dans la molécule) où l’on
retrouve les harmoniques des bandes de déformation hors du plan et dans le plan
: c’est la signature de la molécule aromatique, qui peut confirmer, si
nécessaire, les informations obtenues grâce aux ![]() . Il faut aussi
rappeler les bandes
. Il faut aussi
rappeler les bandes ![]() (un peu
au dessus de 3000
(un peu
au dessus de 3000 ![]() , ici 3008
, ici 3008 ![]() ), avec un plus
grand nombre de bandes pour l’alcool benzylique, entre 3100 et 3000
), avec un plus
grand nombre de bandes pour l’alcool benzylique, entre 3100 et 3000 ![]() (B).
Il existe également plusieurs modes d’élongation des liaisons C = C aromatiques
: dans cet exemple ils apparaissent à 1605, 1495, 1466
(B).
Il existe également plusieurs modes d’élongation des liaisons C = C aromatiques
: dans cet exemple ils apparaissent à 1605, 1495, 1466 ![]() . S’il y a
conjugaison du cycle avec un doublet p ou s non liant, il peut
apparaître une quatrième bande.
. S’il y a
conjugaison du cycle avec un doublet p ou s non liant, il peut
apparaître une quatrième bande.
Les bandes caractéristiques concernent les
liaisons C–O et O–H . L’élongation de O–H d’un alcool donne une absorption
intense dont la fréquence dépend de l’existence ou non de liaisons hydrogène :
![]() . Pour une molécule diluée dans un solvant aprotique apolaire,
donc lorsqu’il n’y a pas de liaisons H, la fréquence
. Pour une molécule diluée dans un solvant aprotique apolaire,
donc lorsqu’il n’y a pas de liaisons H, la fréquence ![]() se situe entre
3600 et 3584
se situe entre
3600 et 3584 ![]() . Par contre pour l’alcool benzylique pur, avec de fortes et
nombreuses liaisons H , cette fréquence descend à 3300
. Par contre pour l’alcool benzylique pur, avec de fortes et
nombreuses liaisons H , cette fréquence descend à 3300 ![]() . L’alcool
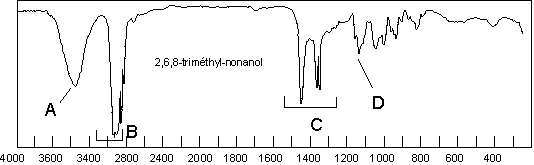
secondaire suivant (2,6,8-triméthyl-nonan-4-ol) voit son
. L’alcool
secondaire suivant (2,6,8-triméthyl-nonan-4-ol) voit son ![]() à 3355
à 3355 ![]() . Laissons de
côté les bandes déjà étudiées (
. Laissons de
côté les bandes déjà étudiées (![]() ,
, ![]() ,
, ![]() ). Selon le
type d’alcool (primaire, secondaire ou tertiaire), les
). Selon le
type d’alcool (primaire, secondaire ou tertiaire), les ![]() et
et ![]() auront des absorptions différentes :
auront des absorptions différentes :
|
I |
II |
III |
phénol | |
|
|
1208 |
1355 |
vers 1380 |
1360 |
|
|
1017 |
1138 |
vers 1160 |
1223 |

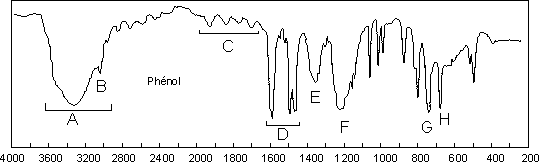
Le phénol montre tous les pics précédents, avec
les effets de la conjugaison entre les électrons p du cycle et le doublet non
liant de O : hyperchrome en général, hypsochrome pour ![]() (3045
(3045 ![]() ),
), ![]() (1360
(1360 ![]() ) et
) et ![]() (1223
(1223
![]() ), et bathochrome pour
), et bathochrome pour ![]() aromatique (1580
aromatique (1580 ![]() en particulier). Les deux bandes
en particulier). Les deux bandes ![]() pour la monosubstitution se retrouvent à 685 et 745
pour la monosubstitution se retrouvent à 685 et 745 ![]() (G et
H).
(G et
H).
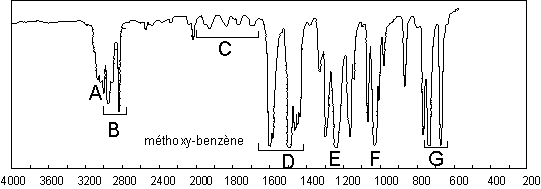
La réponse caractéristique des éthers est associée
à l’élongation du système C–O–C. Il y a une bande d’élongation symétrique
(faible en général, sauf s’il y a conjugaison) : 1030 ![]() pour l’anisole
; et une bande d’élongation asymétrique, toujours forte, vers 1200
pour l’anisole
; et une bande d’élongation asymétrique, toujours forte, vers 1200 ![]() (1245
(1245 ![]() pour l’anisole : E)
pour l’anisole : E)
Tous les composés organiques comportant un
groupement carbonyle C=O ont une absorption caractéristique intense vers 1700
![]() : c’est la bande la plus intense et la plus nette d’un spectre IR. La valeur de
l’absorption du C=O dépend de l’état physique (solide, liquide, vapeur, en
solution) , des effets dus aux groupes voisins, de la conjugaison, et des
liaisons H éventuelles.
: c’est la bande la plus intense et la plus nette d’un spectre IR. La valeur de
l’absorption du C=O dépend de l’état physique (solide, liquide, vapeur, en
solution) , des effets dus aux groupes voisins, de la conjugaison, et des
liaisons H éventuelles.
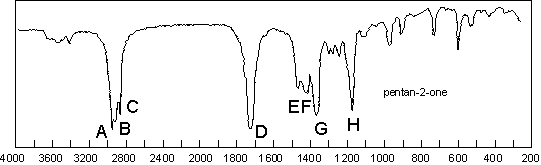
Une cétone aliphatique absorbe vers 1715 ![]() . Le
remplacement d’un groupement saturé par un hétéroatome provoque un effet
hypsochrome si l'effet –I prédomine (–X, mais aussi –O d’un ester, acide,
anhydride,...) et un effet bathochrome si l’effet +E prédomine (–N, –S,...). La
conjugaison avec une double liaison C=C diminue la force de la liaison C=O et de
la liaison C=C. Il y a effet bathochrome pour les deux absorptions
. Le
remplacement d’un groupement saturé par un hétéroatome provoque un effet
hypsochrome si l'effet –I prédomine (–X, mais aussi –O d’un ester, acide,
anhydride,...) et un effet bathochrome si l’effet +E prédomine (–N, –S,...). La
conjugaison avec une double liaison C=C diminue la force de la liaison C=O et de
la liaison C=C. Il y a effet bathochrome pour les deux absorptions ![]() et
et
![]() (1685 -1666
(1685 -1666 ![]() pour le
pour le ![]() ). La conjugaison ne se
fait pas sentir pour les a -dicétones R–CO–CO–R.
). La conjugaison ne se
fait pas sentir pour les a -dicétones R–CO–CO–R.
Sur les deux spectres de cétones proposés, on va
retrouver les ![]() respectivement à 1725
respectivement à 1725 ![]() (non conjugué)
et 1683
(non conjugué)
et 1683 ![]() (conjugué). Il faut remarquer l’existence d’une bande
d’élongation C–CO–C, de faible intensité, à 1172
(conjugué). Il faut remarquer l’existence d’une bande
d’élongation C–CO–C, de faible intensité, à 1172 ![]() pour le
premier composé, à 1255
pour le
premier composé, à 1255 ![]() , plus forte, pour la cétone aromatique. Cette bande
est à distinguer de celle des esters et des acides (beaucoup plus forte, dans la
même zone de nombre d’onde).
, plus forte, pour la cétone aromatique. Cette bande
est à distinguer de celle des esters et des acides (beaucoup plus forte, dans la
même zone de nombre d’onde).
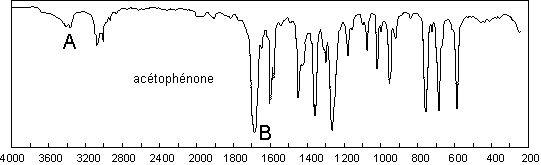
Les contraintes dues aux cycles ont un effet
hypsochrome sur le ![]() . Ainsi la cyclohexanone absorbe-t-elle à 1715
. Ainsi la cyclohexanone absorbe-t-elle à 1715 ![]() , la
cyclopentanone à 1751
, la
cyclopentanone à 1751 ![]() et la cyclobutanone à 1775
et la cyclobutanone à 1775 ![]() .
.

L’absorption de ![]() se fait pour
une fréquence un peu plus élevée que pour une cétone (1740–1720
se fait pour
une fréquence un peu plus élevée que pour une cétone (1740–1720 ![]() ). On retrouve
l’influence des effets –I et +E, ainsi que celle de la conjugaison. Le
trichloroéthanal absorbe ainsi à 1768
). On retrouve
l’influence des effets –I et +E, ainsi que celle de la conjugaison. Le
trichloroéthanal absorbe ainsi à 1768 ![]() . De nouvelles
bandes apparaissent, celles dues à l’absorption
. De nouvelles
bandes apparaissent, celles dues à l’absorption ![]() aldéhydique. Le premier sort sous forme d’un doublet (C)
(ici 2825 et 2717
aldéhydique. Le premier sort sous forme d’un doublet (C)
(ici 2825 et 2717 ![]() ) au dessous des
) au dessous des ![]() aliphatiques. Le second sort à 1389
aliphatiques. Le second sort à 1389 ![]() (F) (peu
important).
(F) (peu
important).
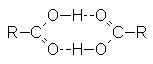
Deux bandes importantes : ![]() et
et ![]() , et deux
bandes mineures :
, et deux
bandes mineures : ![]() et . Les acides
carboxyliques existent sous forme de dimères à cause des très fortes liaisons H
existant entre O–H et C=O :
et . Les acides
carboxyliques existent sous forme de dimères à cause des très fortes liaisons H
existant entre O–H et C=O :
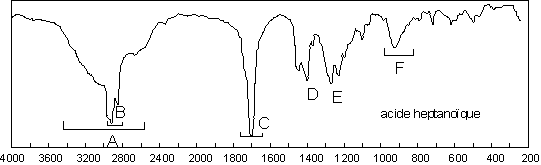
Ainsi observe-t-on la plupart du temps le ![]() du
dimère. En solution très diluée dans un solvant apolaire,
du
dimère. En solution très diluée dans un solvant apolaire, ![]() vaut 3520
vaut 3520 ![]() .
Lorsque le dimère existe, on a au contraire une bande très large et très intense
entre 3300 et 2500
.
Lorsque le dimère existe, on a au contraire une bande très large et très intense
entre 3300 et 2500 ![]() , sur laquelle se superposent les
, sur laquelle se superposent les ![]() alkyles et aryles (B sur le spectre de l’acide
heptanoïque). Le du
alkyles et aryles (B sur le spectre de l’acide
heptanoïque). Le du ![]() du monomère est intense
et absorbe vers 1760
du monomère est intense
et absorbe vers 1760 ![]() (effet –I de O qui prime ici). Dans le dimère (qui est
la structure habituelle), la liaison C=O est affaiblie par la liaison H et la
bande d’absorption subit un effet bathochrome important : entre 1720 et 1706
(effet –I de O qui prime ici). Dans le dimère (qui est
la structure habituelle), la liaison C=O est affaiblie par la liaison H et la
bande d’absorption subit un effet bathochrome important : entre 1720 et 1706
![]() (ici 1715
(ici 1715 ![]() ). Les effets électroniques sont toujours à prendre en compte. À
1408
). Les effets électroniques sont toujours à prendre en compte. À
1408 ![]() , on trouve
, on trouve ![]() , à 1280
, à 1280 ![]() ,
, ![]() ; à 930
; à 930 ![]() ,
,
![]() .
.
On trouve deux bandes pour ![]() , avec un
effet bathochrome par rapport à la bande C=O : 1600
, avec un
effet bathochrome par rapport à la bande C=O : 1600 ![]() (symétrique)
(m) et 1385
(symétrique)
(m) et 1385 ![]() (asymétrique) (F).
(asymétrique) (F).

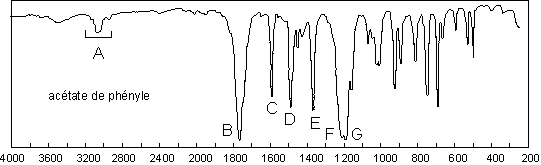
Ceux-ci ont deux bandes intenses qui permettent de
bien les identifier : les ![]() et
et ![]() . À cause des
effets –I de O (tempérés ici par les effets +I du groupe alkyle), l’absorption
. À cause des
effets –I de O (tempérés ici par les effets +I du groupe alkyle), l’absorption
![]() subit un effet
hypsochrome : 1750 – 1735
subit un effet
hypsochrome : 1750 – 1735 ![]() . Si le groupement lié à O est insaturé (comme c’est le
cas ici), la conjugaison du doublet non-liant de O avec la double liaison
dégarnit l’oxygène de quelques pour-cent d’électron ; ceci va augmenter l’effet
–I de O et donc la fréquence d’absorption du
. Si le groupement lié à O est insaturé (comme c’est le
cas ici), la conjugaison du doublet non-liant de O avec la double liaison
dégarnit l’oxygène de quelques pour-cent d’électron ; ceci va augmenter l’effet
–I de O et donc la fréquence d’absorption du ![]() : 1770
: 1770 ![]() pour
l’éthanoate de phényle. L’effet de cycle (lactones) joue comme pour les cétones
cycliques : les g –lactones absorbent à 1795 – 1760
pour
l’éthanoate de phényle. L’effet de cycle (lactones) joue comme pour les cétones
cycliques : les g –lactones absorbent à 1795 – 1760 ![]() . La
conjugaison avec le C=O a comme prévu un effet bathochrome sur
. La
conjugaison avec le C=O a comme prévu un effet bathochrome sur ![]() (1730 – 1715
(1730 – 1715
![]() pour les benzoates).
pour les benzoates).
Il y a deux élongations couplées qui font
intervenir la liaison C–O : ![]() et
et ![]() . La
première est très intense : 1210 – 1260
. La
première est très intense : 1210 – 1260 ![]() (ici 1205). La seconde l’est
surtout pour les esters de phénol : 1030 – 1190
(ici 1205). La seconde l’est
surtout pour les esters de phénol : 1030 – 1190 ![]() (ici 1183).
(ici 1183).
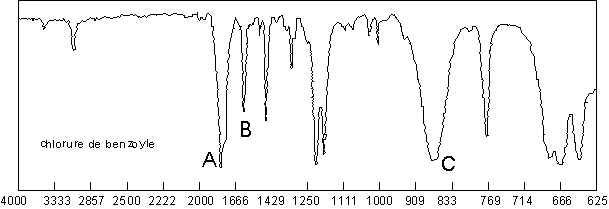
Le C=O subit un effet hypsochrome, entre 1815 et
1785 ![]() (1870 pour
les fluorures). Ici, l’absorption se fait à 1790
(1870 pour
les fluorures). Ici, l’absorption se fait à 1790 ![]() à cause de la conjugaison (A).
Le C–Cl vibre à 875
à cause de la conjugaison (A).
Le C–Cl vibre à 875 ![]() (C), et donne un harmonique assez net à 1745
(C), et donne un harmonique assez net à 1745 ![]() (B).
(B).
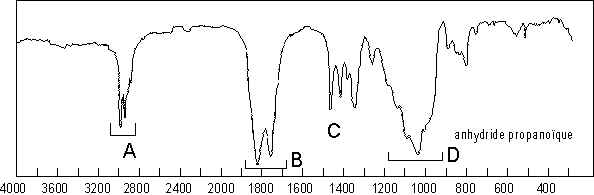
Les deux carbonyles vibrent de manière couplée :
![]() . Aussi
observe-t-on des fréquences d’élongation asymétrique (1825
. Aussi
observe-t-on des fréquences d’élongation asymétrique (1825 ![]() ) et symétrique
(1758
) et symétrique
(1758 ![]() ). À 1040
). À 1040 ![]() , il s’agit de l’élongation
, il s’agit de l’élongation ![]() symétrique et
asymétrique.
symétrique et
asymétrique.
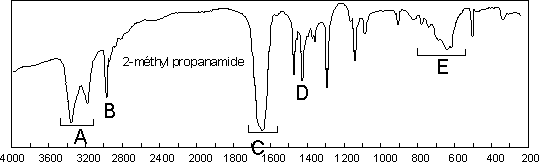
Les amides sont caractérisées par les vibrations
relatives à C=O , N–H essentiellement, C–N accessoirement (![]() = 1425
= 1425 ![]() ). Le
). Le ![]() sort
pour une fréquence plus basse que dans le cas des cétones (effets +E de N) et
recouvre la bande correspondante au
sort
pour une fréquence plus basse que dans le cas des cétones (effets +E de N) et
recouvre la bande correspondante au ![]() (1640
(1640 ![]() pour
ces bandes). Dans le cas de la N’N–diméthyl–méthanamide, seul le
pour
ces bandes). Dans le cas de la N’N–diméthyl–méthanamide, seul le ![]() existe (1680
existe (1680
![]() ). Les bandes de vibration
). Les bandes de vibration ![]() sortent aux
alentours de 3250
sortent aux
alentours de 3250 ![]() dans les produits purs à cause des liaisons H. Il y a
deux bandes pour les amides primaires (élongations symétrique et asymétrique)
(ici 3350 et 3170
dans les produits purs à cause des liaisons H. Il y a
deux bandes pour les amides primaires (élongations symétrique et asymétrique)
(ici 3350 et 3170 ![]() ), l’asymétrique étant la plus intense. On ne trouve
qu’une bande dans cette zone pour les amides secondaires (3210
), l’asymétrique étant la plus intense. On ne trouve
qu’une bande dans cette zone pour les amides secondaires (3210 ![]() pour la
N–éthylpropanamide), et pas de bande du tout pour les amides tertiaires. À
remarquer encore la bande large
pour la
N–éthylpropanamide), et pas de bande du tout pour les amides tertiaires. À
remarquer encore la bande large ![]() à 700–600
à 700–600 ![]() .
.
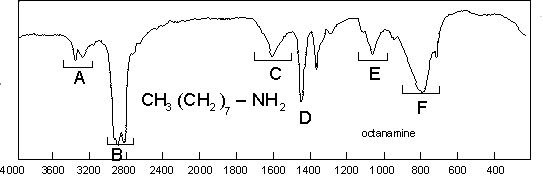
Comme pour les amides, on retrouve, mais en moins
intense, les bandes suivantes : ![]() : deux pour
les amines I (3365 et 3290
: deux pour
les amines I (3365 et 3290 ![]() ici), une pour
les amines II et zéro pour les amines III ;
ici), une pour
les amines II et zéro pour les amines III ; ![]() : 1063
: 1063 ![]() (pas
de conjugaison) ;
(pas
de conjugaison) ;![]() : 1620
: 1620 ![]() et
et ![]() : 910 –660
: 910 –660 ![]() .
.
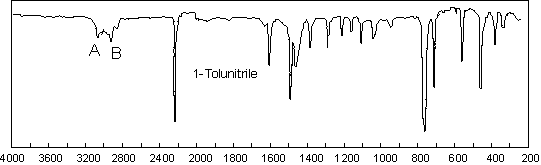
La bande ![]() sort, comme
pour les acétyléniques, vers 2200
sort, comme
pour les acétyléniques, vers 2200 ![]() (2210 ici),
mais elle est plus intense. D’autres groupements absorbent intensément dans
cette zone : les isocyanates –N=C=O , les isothiocyanates –N=C=S , les diimides
–N=C=N– et les isonitriles
(2210 ici),
mais elle est plus intense. D’autres groupements absorbent intensément dans
cette zone : les isocyanates –N=C=O , les isothiocyanates –N=C=S , les diimides
–N=C=N– et les isonitriles ![]() .
.
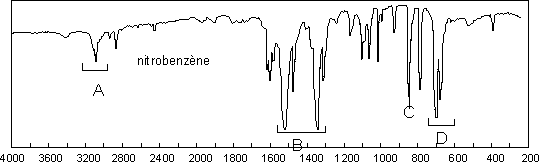
Deux bandes très intenses correspondant aux
élongations asymétrique (1520 ![]() ) et symétrique
(1345
) et symétrique
(1345 ![]() ) du groupement
) du groupement ![]() ressortent
très nettement du spectre. Le
ressortent
très nettement du spectre. Le ![]() est
relativement intense à 850
est
relativement intense à 850 ![]() .
.
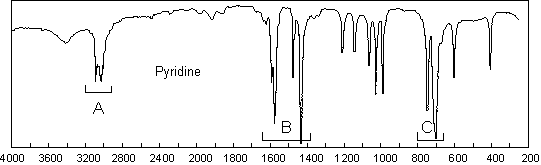
On retrouve les mêmes modes de vibration que pour les aromatiques :
* ![]() , entre
3077 et 3003
, entre
3077 et 3003 ![]() , comme pour les aromatiques (il y a ici un grand nombre de
modes d’élongation)
, comme pour les aromatiques (il y a ici un grand nombre de
modes d’élongation)
* ![]() ; lorsqu’elle
existe, cette liaison fait apparaître une bande entre 3500 et 3220
; lorsqu’elle
existe, cette liaison fait apparaître une bande entre 3500 et 3220 ![]() (cf. amides) . c’est
le cas pour le pyrrole, l’imidazole, l’indole,...
(cf. amides) . c’est
le cas pour le pyrrole, l’imidazole, l’indole,...
* ![]() ; comme dans
le cas des benzènes substitués, on compte le nombre d’atomes d’hydrogène
adjacents pouvant se déformer de manière couplée. Ainsi, pour la pyridine, il y
a 5 H adjacents, ce qui correspond à un benzène monosubstitué, et donc à deux
modes de déformation hors du plan à 748 et 703
; comme dans
le cas des benzènes substitués, on compte le nombre d’atomes d’hydrogène
adjacents pouvant se déformer de manière couplée. Ainsi, pour la pyridine, il y
a 5 H adjacents, ce qui correspond à un benzène monosubstitué, et donc à deux
modes de déformation hors du plan à 748 et 703 ![]() . Il y a 4
bandes de squelette (B) pour la pyridine, moins pour les cycles à 5
chaînons.
. Il y a 4
bandes de squelette (B) pour la pyridine, moins pour les cycles à 5
chaînons.
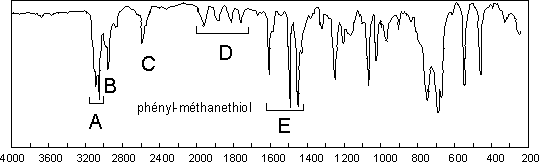
Les Thiols sont remarquables par l’existence d’une
bande assez faible vers 2560 ![]() (ici C : 2665
(ici C : 2665
![]() ). Comme les dérivés nitrés, les sulfones et autres acides
sulfoniques, sulfonates,..., présentent deux bandes très fortes vers 1350
). Comme les dérivés nitrés, les sulfones et autres acides
sulfoniques, sulfonates,..., présentent deux bandes très fortes vers 1350 ![]() (ici
1351
(ici
1351 ![]() ) et vers 1180
) et vers 1180 ![]() (ici 1176
(ici 1176 ![]() ).
).
20. Méthode d’étude d’un spectre IR :
1. Rechercher la présence d’un groupe C=O : présence d’une bande intense vers 1700 - 1800 cm–1. Si oui, continuer ci-dessous, sinon, passer au §2
1.1. Essayer de trouver d’autres bandes caractéristiques des fonctions comprenant un C=O :
· doublet![]() des
aldéhydes entre 2650 et 2800 cm–1.
des
aldéhydes entre 2650 et 2800 cm–1.
· bande large et
forte![]() des
acides entre 2500 et 3300 cm–1
des
acides entre 2500 et 3300 cm–1
· bande très
forte![]() des
esters à 1200 cm–1
des
esters à 1200 cm–1
· bande attenante au![]() de
la fonction amide primaire et secondaire :
de
la fonction amide primaire et secondaire :![]() vers
1650 cm–1 et bande(s)
vers
1650 cm–1 et bande(s)![]() vers
3300 cm–1 (F ; 2 bandes pour les primaires et une pour les
secondaires)
vers
3300 cm–1 (F ; 2 bandes pour les primaires et une pour les
secondaires)
1.2. vérifier la fréquence d’absorption du![]() en
fonction des autres bandes
trouvées :
en
fonction des autres bandes
trouvées :
· 1660-1685 pour les amides
· 1700 pour les acides
· 1715 pour les cétones
· 1720-25 pour les aldéhydes
· 1740-55 pour les esters
· 1780-1850 pour les lactones
· 1800-1870 pour les halogénures d’acide
passer au § suivant
2. Rechercher la présence de bandes fortes
et pas trop larges vers 3250 – 3500 cm–1. Il s’agit d’élongations![]() des
alcools (TF ; 3350) ,
des
alcools (TF ; 3350) ,![]() des
amines (mf ; 2 bandes pour les primaires et une pour les secondaires) ,
des
amines (mf ; 2 bandes pour les primaires et une pour les secondaires) ,![]() des
alcynes vrais (F à TF, vers 3250).
des
alcynes vrais (F à TF, vers 3250).
3. Il reste les fonctions particulières :
· dérivés halogénés
· dérivés soufrés
· dérivés azotés (nitriles, isocyanates, etc...)
pour tous ceux-là, voir le tableau des valeurs IR
4. Enfin, étude des liaisons C–H autres que celles vues auparavant :
· ![]() :
alcanes : 2850 à 2950 cm–1
:
alcanes : 2850 à 2950 cm–1
· ![]() :
alcènes : 3050 à 3080 cm–1, avec les
:
alcènes : 3050 à 3080 cm–1, avec les![]() à
1640 cm–1 (v. aussi les
à
1640 cm–1 (v. aussi les![]()
· ![]() :
aromatiques : 3020 à 3050 cm–1, avec les
:
aromatiques : 3020 à 3050 cm–1, avec les![]() caractéristiques
de la substitution (voir tableau) vers 650 - 900 cm–1, et les
caractéristiques
de la substitution (voir tableau) vers 650 - 900 cm–1, et les![]() vers
1450 – 1600 .
vers
1450 – 1600 .
