Spectroscopie infrarouge
D'apr�s " SPECTROMETRIC IDENDIFICATION OF ORGANIC COMPOUNDS"
par SILVERSTEIN, BASSLER et MORILL. Editeur : John Wiley & Sons.
Pour plus de renseignements vous pouvez contacter
Jean Umber,
Professeur de Chimie au Lyc�e Louis Vincent � Metz (57000).
Liens :
>> Voir aussi cours de chimie Page d'accueil
Les transitions �nerg�tiques se font ici entre les niveaux d’�nergie de rotation des mol�cules ou entre leurs niveaux d’�nergie de vibration.
Les transitions entre niveaux de rotation apparaissent dans
l’I.R. lointain (de 20 � 250 mm
ou de 500 � 40 ![]() ). Les transitions entre niveaux vibrationnels
apparaissent de 1 � 20 mm (ou de 10
000 � 500
). Les transitions entre niveaux vibrationnels
apparaissent de 1 � 20 mm (ou de 10
000 � 500 ![]() ). Nous ferons porter notre �tude sur les
transitions vibrationnelles. On constate qu’elles n�cessitent plus d’�nergie
que les transitions rotationnelles. Aussi la lumi�re excitatrice provoquera-t-elle, pour
chaque transition vibrationnelle, une multitude de transitions rotationnelles, qui vont
donner au pic de transition vibrationnelle l’allure d’une bande
d’absorption :
). Nous ferons porter notre �tude sur les
transitions vibrationnelles. On constate qu’elles n�cessitent plus d’�nergie
que les transitions rotationnelles. Aussi la lumi�re excitatrice provoquera-t-elle, pour
chaque transition vibrationnelle, une multitude de transitions rotationnelles, qui vont
donner au pic de transition vibrationnelle l’allure d’une bande
d’absorption :
1. �longation.
Appel� aussi vibration de valence ou "stretching", ce mode concerne la vibration de la mol�cule le long des liaisons. La fr�quence de vibration est donn�e par la relation :
![]()
o� k est la constante de force de la liaison (consid�r�e ici
comme un ressort), proportionnelle � l’�nergie de liaison, et m la masse r�duite des deux atomes reli�s par
cette liaison. Ainsi, les liaisons multiples, plus �nerg�tiques que les simples, auront
une constante de force plus �lev�e, donc une fr�quence de vibration (remplac�e dans la
pratique par le nombre d’onde) plus �lev�e que celles des liaisons simples entre
atomes identiques : C–C absorbe vers 1100 ![]() , C=C vers 1600
, C=C vers 1600 ![]() et C� C vers 2100
et C� C vers 2100 ![]() . Par contre, les liaisons X–H, o� X est un atome quelconque (C, N, O,
...), auront une fr�quence d’�longation plus �lev�e que celle d’une liaison
C–X, car la masse r�duite m y
est plus petite : pour C–H, m =
0,92 u.a. ; pour C–C, m = 6 u.a.
. Par contre, les liaisons X–H, o� X est un atome quelconque (C, N, O,
...), auront une fr�quence d’�longation plus �lev�e que celle d’une liaison
C–X, car la masse r�duite m y
est plus petite : pour C–H, m =
0,92 u.a. ; pour C–C, m = 6 u.a.
2. D�formations dans et hors du plan.
Consid�rons une structure ![]() . En plus de la vibration de valence, l’angle des liaisons peut varier :
il y a flexion ou d�formation. Ces d�formations peuvent avoir lieu dans le plan des deux
liaisons concern�es (on les note d )
ou hors du plan (on les note g ). Il
y a aussi possibilit� de d�formations sym�triques et asym�triques. voici quelques
exemples :
. En plus de la vibration de valence, l’angle des liaisons peut varier :
il y a flexion ou d�formation. Ces d�formations peuvent avoir lieu dans le plan des deux
liaisons concern�es (on les note d )
ou hors du plan (on les note g ). Il
y a aussi possibilit� de d�formations sym�triques et asym�triques. voici quelques
exemples :


Application de l’I.R. � la d�termination des diverses fonctions d’un compos� organique.
Non seulement la nature des deux atomes vibrants intervient dans la valeur de la constante de force, mais aussi l’environnement �lectronique. Aussi chaque groupement fonctionnel aura-t-il des fr�quences caract�ristiques d’�longation et de d�formation. Nous allons passer en revue les diverses fonctions gr�ce � l’�tude de quelques spectres :
1. Les groupements carbon�s satur�s : les alcanes.

On trouve principalement les vibrations d’�longation de la
liaison C–H entre 3000 et 2840 ![]() .
Nous retrouvons ici les fr�quences suivantes :
.
Nous retrouvons ici les fr�quences suivantes :

Il suffira de rep�rer une absorption dans ce domaine pour suspecter fortement la pr�sence de liaisons C–H.
Vers 1400 ![]() se
situent les vibrations de d�formation dans le plan des liaisons C–H :
se
situent les vibrations de d�formation dans le plan des liaisons C–H :
![]()
Une vibration de d�formation hors du plan des ![]() appara�t � 722
appara�t � 722 ![]() . Les
. Les ![]() sont tr�s faibles et se situent entre
1200 et 1800
sont tr�s faibles et se situent entre
1200 et 1800 ![]() .
.
2. Doubles liaisons carbone - carbone.
Par rapport � l’exemple pr�c�dent, il appara�t deux pics
nouveaux : � 1645 ![]() , il s’agit de
, il s’agit de
![]() . � 3050
. � 3050 ![]() , il s’agit
de
, il s’agit
de ![]() . Les vibrations des groupements satur�s apparaissent toujours, et il faut
encore remarquer les deux bandes
. Les vibrations des groupements satur�s apparaissent toujours, et il faut
encore remarquer les deux bandes ![]() � 986 et 907
� 986 et 907 ![]() . Ces deux bandes ne sont � �tudier que
s’il y a un probl�me de st�r�ochimie �thyl�nique (Z ou E) non soluble par
ailleurs.
. Ces deux bandes ne sont � �tudier que
s’il y a un probl�me de st�r�ochimie �thyl�nique (Z ou E) non soluble par
ailleurs.

Lorsque les doubles liaisons sont conjugu�es :

Les trois bandes pr�c�dentes subissent un effet hyperchrome ; le
![]() subit en outre un effet hypsochrome et les autres
subit en outre un effet hypsochrome et les autres
![]() un effet bathochrome :
un effet bathochrome :

3. Triple liaison carbone–carbone.

Il faut remarquer la faible bande de l’�longation
![]() � 2110
� 2110 ![]() . On ne la voit pas
toujours, surtout lorsqu’il s’agit d’alcynes disubstitu�s. Par contre, la
bande d’�longation
. On ne la voit pas
toujours, surtout lorsqu’il s’agit d’alcynes disubstitu�s. Par contre, la
bande d’�longation ![]() des alcynes monosubstitu�s est toujours
intense et sort ici � 3268
des alcynes monosubstitu�s est toujours
intense et sort ici � 3268 ![]() . Moins
importante � signaler est la bande de d�formation de
. Moins
importante � signaler est la bande de d�formation de ![]() ac�tyl�nique (630
ac�tyl�nique (630 ![]() ) , ainsi
que son premier harmonique (1247
) , ainsi
que son premier harmonique (1247 ![]() ).
).
4. Compos�s aromatiques mononucl�aires (benz�no�des).

Il faut toujours s’int�resser aux bandes des basses
fr�quences : de 900 � 650 ![]() . C’est
l� que l’on trouve les renseignements concernant le nombre de substituants du cycle
aromatique et leur position l’un par rapport � l’autre. Sur notre exemple,
l’unique bande de d�formation hors du plan de la liaison
. C’est
l� que l’on trouve les renseignements concernant le nombre de substituants du cycle
aromatique et leur position l’un par rapport � l’autre. Sur notre exemple,
l’unique bande de d�formation hors du plan de la liaison ![]() aromatique
aromatique ![]() montre
l’existence d’une disubstitution –1,2 ; et ce d’autant plus s�rement
qu’il s’agit d’une bande intense.
montre
l’existence d’une disubstitution –1,2 ; et ce d’autant plus s�rement
qu’il s’agit d’une bande intense.
Dans l’exemple suivant (alcool benzylique avec cycle
monosubstitu�), on trouve deux bandes fortes ![]() , correspondant aux
deux modes privil�gi�s de d�formation hors du plan pour 5 hydrog�nes aromatiques
adjacents. On trouve, dans la zone allant de 1300 � 1000
, correspondant aux
deux modes privil�gi�s de d�formation hors du plan pour 5 hydrog�nes aromatiques
adjacents. On trouve, dans la zone allant de 1300 � 1000 ![]() les bandes de d�formation dans le plan des H aromatiques. Elles
sont plut�t faibles et nous ne nous en serviront pas pour la d�termination
fonctionnelle.
les bandes de d�formation dans le plan des H aromatiques. Elles
sont plut�t faibles et nous ne nous en serviront pas pour la d�termination
fonctionnelle.
Int�ressante aussi est la zone comprise entre 2000 et 1667 ![]() (lorsqu’il n’y a pas de carbonyle
dans la mol�cule) o� l’on retrouve les harmoniques des bandes de d�formation hors
du plan et dans le plan : c’est la signature de la mol�cule aromatique, qui peut
confirmer, si n�cessaire, les informations obtenues gr�ce aux
(lorsqu’il n’y a pas de carbonyle
dans la mol�cule) o� l’on retrouve les harmoniques des bandes de d�formation hors
du plan et dans le plan : c’est la signature de la mol�cule aromatique, qui peut
confirmer, si n�cessaire, les informations obtenues gr�ce aux ![]() . Il faut aussi rappeler les bandes
. Il faut aussi rappeler les bandes ![]() (un peu au dessus de 3000
(un peu au dessus de 3000 ![]() , ici 3008
, ici 3008 ![]() ), avec un plus grand nombre de bandes pour
l’alcool benzylique, entre 3100 et 3000
), avec un plus grand nombre de bandes pour
l’alcool benzylique, entre 3100 et 3000 ![]() (B). Il existe �galement plusieurs modes d’�longation des liaisons C =
C aromatiques : dans cet exemple ils apparaissent � 1605, 1495, 1466
(B). Il existe �galement plusieurs modes d’�longation des liaisons C =
C aromatiques : dans cet exemple ils apparaissent � 1605, 1495, 1466 ![]() . S’il y a conjugaison du cycle avec un
doublet p ou s non liant, il peut appara�tre une quatri�me
bande.
. S’il y a conjugaison du cycle avec un
doublet p ou s non liant, il peut appara�tre une quatri�me
bande.
Les bandes caract�ristiques concernent les liaisons C–O et
O–H . L’�longation de O–H d’un alcool donne une absorption intense
dont la fr�quence d�pend de l’existence ou non de liaisons hydrog�ne : ![]() . Pour une mol�cule dilu�e dans un solvant
aprotique apolaire, donc lorsqu’il n’y a pas de liaisons H, la fr�quence
. Pour une mol�cule dilu�e dans un solvant
aprotique apolaire, donc lorsqu’il n’y a pas de liaisons H, la fr�quence ![]() se situe entre 3600 et 3584
se situe entre 3600 et 3584 ![]() . Par contre pour l’alcool benzylique pur,
avec de fortes et nombreuses liaisons H , cette fr�quence descend � 3300
. Par contre pour l’alcool benzylique pur,
avec de fortes et nombreuses liaisons H , cette fr�quence descend � 3300 ![]() . L’alcool secondaire suivant
(2,6,8-trim�thyl-nonan-4-ol) voit son
. L’alcool secondaire suivant
(2,6,8-trim�thyl-nonan-4-ol) voit son ![]() � 3355
� 3355 ![]() . Laissons de c�t� les bandes d�j� �tudi�es (
. Laissons de c�t� les bandes d�j� �tudi�es (![]() ,
, ![]() ,
, ![]() ). Selon le type d’alcool (primaire,
secondaire ou tertiaire), les
). Selon le type d’alcool (primaire,
secondaire ou tertiaire), les ![]() et
et ![]() auront des absorptions diff�rentes :
auront des absorptions diff�rentes :
I |
II |
III |
ph�nol |
|
|
1208 |
1355 |
vers 1380 |
1360 |
|
1017 |
1138 |
vers 1160 |
1223 |


Le ph�nol montre tous les pics pr�c�dents, avec les effets de la
conjugaison entre les �lectrons p du
cycle et le doublet non liant de O : hyperchrome en g�n�ral, hypsochrome pour ![]() (3045
(3045 ![]() ),
), ![]() (1360
(1360 ![]() ) et
) et ![]() (1223
(1223 ![]() ), et bathochrome pour
), et bathochrome pour ![]() aromatique (1580
aromatique (1580 ![]() en particulier). Les deux bandes
en particulier). Les deux bandes ![]() pour la monosubstitution se retrouvent � 685 et 745
pour la monosubstitution se retrouvent � 685 et 745 ![]() (G et H).
(G et H).


La r�ponse caract�ristique des �thers est associ�e �
l’�longation du syst�me C–O–C. Il y a une bande d’�longation
sym�trique (faible en g�n�ral, sauf s’il y a conjugaison) : 1030 ![]() pour l’anisole ; et une bande
d’�longation asym�trique, toujours forte, vers 1200
pour l’anisole ; et une bande
d’�longation asym�trique, toujours forte, vers 1200 ![]() (1245
(1245 ![]() pour
l’anisole : E)
pour
l’anisole : E)
Tous les compos�s organiques comportant un groupement carbonyle C=O
ont une absorption caract�ristique intense vers 1700 ![]() : c’est la bande la plus intense et la plus nette d’un spectre IR.
La valeur de l’absorption du C=O d�pend de l’�tat physique (solide, liquide,
vapeur, en solution) , des effets dus aux groupes voisins, de la conjugaison, et des
liaisons H �ventuelles.
: c’est la bande la plus intense et la plus nette d’un spectre IR.
La valeur de l’absorption du C=O d�pend de l’�tat physique (solide, liquide,
vapeur, en solution) , des effets dus aux groupes voisins, de la conjugaison, et des
liaisons H �ventuelles.

Une c�tone aliphatique absorbe vers 1715 ![]() . Le remplacement d’un groupement satur� par un h�t�roatome
provoque un effet hypsochrome si l'effet –I pr�domine (–X, mais aussi –O
d’un ester, acide, anhydride,...) et un effet bathochrome si l’effet +E
pr�domine (–N, –S,...). La conjugaison avec une double liaison C=C diminue la
force de la liaison C=O et de la liaison C=C. Il y a effet bathochrome pour les deux
absorptions
. Le remplacement d’un groupement satur� par un h�t�roatome
provoque un effet hypsochrome si l'effet –I pr�domine (–X, mais aussi –O
d’un ester, acide, anhydride,...) et un effet bathochrome si l’effet +E
pr�domine (–N, –S,...). La conjugaison avec une double liaison C=C diminue la
force de la liaison C=O et de la liaison C=C. Il y a effet bathochrome pour les deux
absorptions ![]() et
et ![]() (1685 -1666
(1685 -1666 ![]() pour
le
pour
le ![]() ). La conjugaison ne se fait pas sentir pour les
a -dic�tones R–CO–CO–R.
). La conjugaison ne se fait pas sentir pour les
a -dic�tones R–CO–CO–R.
Sur les deux spectres de c�tones propos�s, on va retrouver les ![]() respectivement � 1725
respectivement � 1725 ![]() (non conjugu�) et 1683
(non conjugu�) et 1683 ![]() (conjugu�). Il faut remarquer
l’existence d’une bande d’�longation C–CO–C, de faible
intensit�, � 1172
(conjugu�). Il faut remarquer
l’existence d’une bande d’�longation C–CO–C, de faible
intensit�, � 1172 ![]() pour le premier
compos�, � 1255
pour le premier
compos�, � 1255 ![]() , plus forte, pour la
c�tone aromatique. Cette bande est � distinguer de celle des esters et des acides
(beaucoup plus forte, dans la m�me zone de nombre d’onde).
, plus forte, pour la
c�tone aromatique. Cette bande est � distinguer de celle des esters et des acides
(beaucoup plus forte, dans la m�me zone de nombre d’onde).

Les contraintes dues aux cycles ont un effet hypsochrome sur le ![]() . Ainsi la cyclohexanone absorbe-t-elle � 1715
. Ainsi la cyclohexanone absorbe-t-elle � 1715
![]() , la cyclopentanone � 1751
, la cyclopentanone � 1751 ![]() et la cyclobutanone � 1775
et la cyclobutanone � 1775 ![]() .
.

L’absorption de ![]() se fait pour une fr�quence un peu plus �lev�e que pour une c�tone (1740–1720
se fait pour une fr�quence un peu plus �lev�e que pour une c�tone (1740–1720 ![]() ). On retrouve l’influence des effets
–I et +E, ainsi que celle de la conjugaison. Le trichloro�thanal absorbe ainsi �
1768
). On retrouve l’influence des effets
–I et +E, ainsi que celle de la conjugaison. Le trichloro�thanal absorbe ainsi �
1768 ![]() . De nouvelles bandes apparaissent,
celles dues � l’absorption
. De nouvelles bandes apparaissent,
celles dues � l’absorption ![]() ald�hydique. Le
premier sort sous forme d’un doublet (C) (ici 2825 et 2717
ald�hydique. Le
premier sort sous forme d’un doublet (C) (ici 2825 et 2717 ![]() ) au dessous des
) au dessous des ![]() aliphatiques. Le
second sort � 1389
aliphatiques. Le
second sort � 1389 ![]() (F) (peu important).
(F) (peu important).
Deux bandes importantes : ![]() et
et ![]() , et deux bandes mineures :
, et deux bandes mineures : ![]() et . Les acides carboxyliques existent sous forme de dim�res � cause des tr�s
fortes liaisons H existant entre O–H et C=O :
et . Les acides carboxyliques existent sous forme de dim�res � cause des tr�s
fortes liaisons H existant entre O–H et C=O :
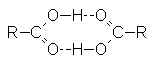
Ainsi observe-t-on la plupart du temps le ![]() du dim�re. En solution tr�s dilu�e dans un solvant apolaire,
du dim�re. En solution tr�s dilu�e dans un solvant apolaire, ![]() vaut 3520
vaut 3520 ![]() . Lorsque le dim�re existe, on a au contraire une bande tr�s large et tr�s
intense entre 3300 et 2500
. Lorsque le dim�re existe, on a au contraire une bande tr�s large et tr�s
intense entre 3300 et 2500 ![]() , sur laquelle
se superposent les
, sur laquelle
se superposent les ![]() alkyles et aryles (B sur le spectre de
l’acide heptano�que). Le du
alkyles et aryles (B sur le spectre de
l’acide heptano�que). Le du ![]() du monom�re est intense et absorbe vers
1760
du monom�re est intense et absorbe vers
1760 ![]() (effet –I de O qui prime ici).
Dans le dim�re (qui est la structure habituelle), la liaison C=O est affaiblie par la
liaison H et la bande d’absorption subit un effet bathochrome important : entre 1720
et 1706
(effet –I de O qui prime ici).
Dans le dim�re (qui est la structure habituelle), la liaison C=O est affaiblie par la
liaison H et la bande d’absorption subit un effet bathochrome important : entre 1720
et 1706 ![]() (ici 1715
(ici 1715 ![]() ). Les effets �lectroniques sont toujours �
prendre en compte. � 1408
). Les effets �lectroniques sont toujours �
prendre en compte. � 1408 ![]() , on trouve
, on trouve ![]() , � 1280
, � 1280 ![]() ,
, ![]() ; � 930
; � 930 ![]() ,
, ![]() .
.

On trouve deux bandes pour ![]() , avec un effet bathochrome par rapport � la bande C=O : 1600
, avec un effet bathochrome par rapport � la bande C=O : 1600 ![]() (sym�trique) (m) et 1385
(sym�trique) (m) et 1385 ![]() (asym�trique) (F).
(asym�trique) (F).


Ceux-ci ont deux bandes intenses qui permettent de bien les
identifier : les ![]() et
et ![]() . � cause des effets –I de O (temp�r�s
ici par les effets +I du groupe alkyle), l’absorption
. � cause des effets –I de O (temp�r�s
ici par les effets +I du groupe alkyle), l’absorption ![]() subit un effet hypsochrome : 1750 – 1735
subit un effet hypsochrome : 1750 – 1735 ![]() . Si le groupement li� � O est insatur�
(comme c’est le cas ici), la conjugaison du doublet non-liant de O avec la double
liaison d�garnit l’oxyg�ne de quelques pour-cent d’�lectron ; ceci va
augmenter l’effet –I de O et donc la fr�quence d’absorption du
. Si le groupement li� � O est insatur�
(comme c’est le cas ici), la conjugaison du doublet non-liant de O avec la double
liaison d�garnit l’oxyg�ne de quelques pour-cent d’�lectron ; ceci va
augmenter l’effet –I de O et donc la fr�quence d’absorption du ![]() : 1770
: 1770 ![]() pour l’�thanoate de ph�nyle. L’effet de cycle (lactones) joue
comme pour les c�tones cycliques : les g –lactones absorbent � 1795 – 1760
pour l’�thanoate de ph�nyle. L’effet de cycle (lactones) joue
comme pour les c�tones cycliques : les g –lactones absorbent � 1795 – 1760 ![]() . La conjugaison avec le C=O a comme pr�vu un effet bathochrome
sur
. La conjugaison avec le C=O a comme pr�vu un effet bathochrome
sur ![]() (1730 – 1715
(1730 – 1715 ![]() pour les benzoates).
pour les benzoates).
Il y a deux �longations coupl�es qui font intervenir la liaison
C–O : ![]() et
et ![]() . La premi�re est tr�s intense : 1210 – 1260
. La premi�re est tr�s intense : 1210 – 1260 ![]() (ici 1205). La seconde l’est surtout pour
les esters de ph�nol : 1030 – 1190
(ici 1205). La seconde l’est surtout pour
les esters de ph�nol : 1030 – 1190 ![]() (ici 1183).
(ici 1183).
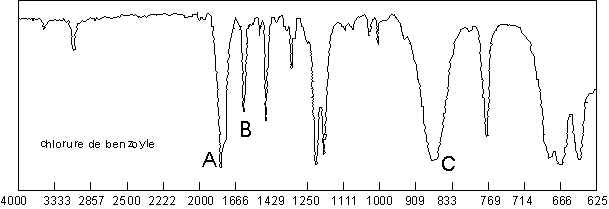
Le C=O subit un effet hypsochrome, entre 1815 et 1785 ![]() (1870 pour les fluorures). Ici,
l’absorption se fait � 1790
(1870 pour les fluorures). Ici,
l’absorption se fait � 1790 ![]() �
cause de la conjugaison (A). Le C–Cl vibre � 875
�
cause de la conjugaison (A). Le C–Cl vibre � 875 ![]() (C), et donne un harmonique assez net � 1745
(C), et donne un harmonique assez net � 1745 ![]() (B).
(B).
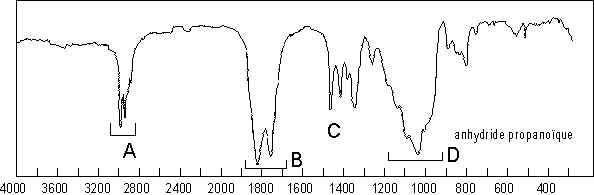
Les deux carbonyles vibrent de mani�re coupl�e : ![]() . Aussi observe-t-on des fr�quences
d’�longation asym�trique (1825
. Aussi observe-t-on des fr�quences
d’�longation asym�trique (1825 ![]() ) et
sym�trique (1758
) et
sym�trique (1758 ![]() ). � 1040
). � 1040 ![]() , il s’agit de l’�longation
, il s’agit de l’�longation ![]() sym�trique et asym�trique.
sym�trique et asym�trique.

Les amides sont caract�ris�es par les vibrations relatives � C=O
, N–H essentiellement, C–N accessoirement (![]() = 1425
= 1425 ![]() ). Le
). Le ![]() sort pour une fr�quence plus basse que dans le cas des c�tones (effets +E
de N) et recouvre la bande correspondante au
sort pour une fr�quence plus basse que dans le cas des c�tones (effets +E
de N) et recouvre la bande correspondante au ![]() (1640
(1640 ![]() pour ces bandes). Dans
le cas de la N’N–dim�thyl–m�thanamide, seul le
pour ces bandes). Dans
le cas de la N’N–dim�thyl–m�thanamide, seul le ![]() existe (1680
existe (1680 ![]() ).
Les bandes de vibration
).
Les bandes de vibration ![]() sortent aux
alentours de 3250
sortent aux
alentours de 3250 ![]() dans les produits purs
� cause des liaisons H. Il y a deux bandes pour les amides primaires (�longations
sym�trique et asym�trique) (ici 3350 et 3170
dans les produits purs
� cause des liaisons H. Il y a deux bandes pour les amides primaires (�longations
sym�trique et asym�trique) (ici 3350 et 3170 ![]() ), l’asym�trique �tant la plus intense. On ne trouve qu’une bande
dans cette zone pour les amides secondaires (3210
), l’asym�trique �tant la plus intense. On ne trouve qu’une bande
dans cette zone pour les amides secondaires (3210 ![]() pour la N–�thylpropanamide), et pas de bande du tout pour les amides
tertiaires. � remarquer encore la bande large
pour la N–�thylpropanamide), et pas de bande du tout pour les amides
tertiaires. � remarquer encore la bande large ![]() � 700–600
� 700–600 ![]() .
.

Comme pour les amides, on retrouve, mais en moins intense, les
bandes suivantes : ![]() : deux pour les amines
I (3365 et 3290
: deux pour les amines
I (3365 et 3290 ![]() ici), une pour les amines
II et z�ro pour les amines III ;
ici), une pour les amines
II et z�ro pour les amines III ; ![]() : 1063
: 1063 ![]() (pas de conjugaison) ;
(pas de conjugaison) ;![]() : 1620
: 1620 ![]() et
et ![]() : 910 –660
: 910 –660 ![]() .
.

La bande ![]() sort,
comme pour les ac�tyl�niques, vers 2200
sort,
comme pour les ac�tyl�niques, vers 2200 ![]() (2210 ici), mais elle est plus intense. D’autres groupements absorbent intens�ment
dans cette zone : les isocyanates –N=C=O , les isothiocyanates –N=C=S , les
diimides –N=C=N– et les isonitriles
(2210 ici), mais elle est plus intense. D’autres groupements absorbent intens�ment
dans cette zone : les isocyanates –N=C=O , les isothiocyanates –N=C=S , les
diimides –N=C=N– et les isonitriles ![]() .
.

Deux bandes tr�s intenses correspondant aux �longations
asym�trique (1520 ![]() ) et sym�trique (1345
) et sym�trique (1345 ![]() ) du groupement
) du groupement ![]() ressortent tr�s nettement du spectre. Le
ressortent tr�s nettement du spectre. Le ![]() est relativement intense � 850
est relativement intense � 850 ![]() .
.

On retrouve les m�mes modes de vibration que pour les aromatiques :
* ![]() , entre 3077 et 3003
, entre 3077 et 3003 ![]() , comme pour les aromatiques (il y a ici un
grand nombre de modes d’�longation)
, comme pour les aromatiques (il y a ici un
grand nombre de modes d’�longation)
* ![]() ;
lorsqu’elle existe, cette liaison fait appara�tre une bande entre 3500 et 3220
;
lorsqu’elle existe, cette liaison fait appara�tre une bande entre 3500 et 3220 ![]() (cf. amides) . c’est le cas pour le
pyrrole, l’imidazole, l’indole,...
(cf. amides) . c’est le cas pour le
pyrrole, l’imidazole, l’indole,...
* ![]() ; comme dans le
cas des benz�nes substitu�s, on compte le nombre d’atomes d’hydrog�ne
adjacents pouvant se d�former de mani�re coupl�e. Ainsi, pour la pyridine, il y a 5 H
adjacents, ce qui correspond � un benz�ne monosubstitu�, et donc � deux modes de
d�formation hors du plan � 748 et 703
; comme dans le
cas des benz�nes substitu�s, on compte le nombre d’atomes d’hydrog�ne
adjacents pouvant se d�former de mani�re coupl�e. Ainsi, pour la pyridine, il y a 5 H
adjacents, ce qui correspond � un benz�ne monosubstitu�, et donc � deux modes de
d�formation hors du plan � 748 et 703 ![]() .
Il y a 4 bandes de squelette (B) pour la pyridine, moins pour les cycles � 5 cha�nons.
.
Il y a 4 bandes de squelette (B) pour la pyridine, moins pour les cycles � 5 cha�nons.
Les Thiols sont remarquables par l’existence d’une bande
assez faible vers 2560 ![]() (ici C : 2665
(ici C : 2665 ![]() ). Comme les d�riv�s nitr�s, les sulfones et
autres acides sulfoniques, sulfonates,..., pr�sentent deux bandes tr�s fortes vers 1350
). Comme les d�riv�s nitr�s, les sulfones et
autres acides sulfoniques, sulfonates,..., pr�sentent deux bandes tr�s fortes vers 1350 ![]() (ici 1351
(ici 1351 ![]() ) et vers 1180
) et vers 1180 ![]() (ici 1176
(ici 1176 ![]() ).
).

